Ginecología y Obstetricia
La especialidadArtículos especializados
Enfermedades
Síntomas
Otros contenidos

Artículo especializado
Resultados de test prenatales: ¿Cuánto tardan y cómo leerlos?
El test prenatal no invasivo es una prueba que permite comprobar el estado del bebé antes de su nacimiento e identificar posibles anomalías genéticas, así como conocer el sexo fetal. Esta prueba se realiza a partir de una muestra de sangre de la madre y sus resultados están disponibles en unos días.
Test prenatales no invasivos
La técnica de los test prenatales se basa en analizar el ADN fetal que está presente en la sangre de la madre durante el embarazo. De este modo, a partir de una muestra de sangre materna se analizan las posibles anomalías cromosómicas del feto en gestación.
La información genética se organiza dentro de cada célula en los cromosomas. Los seres humanos tenemos 23 pares de cromosomas (dos copias de cada par, es decir 46). Los problemas de salud y de desarrollo aparecen cuando falta o sobra algún cromosoma o una parte de ellos.
En este sentido, los test emplean la última tecnología en secuenciación que compara el ADN fetal respecto al ADN materno para detectar determinadas anomalías con gran precisión y fiabilidad. Además, pueden incluir otras pruebas para la detección de trisomías relacionadas con abortos espontáneos en el primer trimestre e identifican microdeleciones en las que una pérdida de material genético puede causar la aparición de síndromes genéticos importantes.
Trisomías
En las trisomías se produce la presencia de un cromosoma adicional, es decir, 3 copias de un cromosoma en lugar de las 2 copias normales.
Los defectos cromosómicos más habituales en forma de trisomías que pueden detectar los test prenatales son los que aparecen en los cromosomas 21, 18 y 13 (síndrome de Down, Edwards y Patau) y las anomalías en los cromosomas sexuales (X e Y).
Microdeleciones
Las microdeleciones son pequeñas pérdidas de material genético que se asocian con graves problemas del desarrollo en el recién nacido. Se incluyen aquí el síndrome de DiGeorge, el síndrome de deleción 1p36, el síndrome de Cri-du-chat, el síndrome de Angelman y el síndrome de Prader-Willi.
El síndrome por microdeleción más frecuente es el de DiGeorge. En él está ausente un pequeño fragmento del cromosoma 22 (22q11.2), lo que acarrea múltiples alteraciones fetales como defectos cardíacos, anomalías en el paladar y la cara, retraso en el desarrollo e inmunodeficiencia, con infecciones frecuentes.
Triploidía
La triploidía es una alteración cromosómica en la que el feto tiene tres copias de cada uno de los cromosomas en lugar de dos (69 cromosomas en total y no 46). Normalmente se produce cuando dos espermatozoides fertilizan un óvulo, pero también puede ocurrir cuando un óvulo diploide es fertilizado por un espermatozoide, o bien en casos en los que un espermatozoide diploide fertiliza un óvulo.
¿Cómo interpretar resultados test prenatal no invasivo?
El Test Prenatal No Invasivo se basa en tecnología de ultrasecuenciación o Next Generation DNA Sequencing , junto con herramientas avanzadas de bioinformática. El test cuantifica la probabilidad de que esté presente alguna de las alteraciones genéticas más comunes. El resultado ofrece una especificidad y sensibilidad muy elevadas.
La interpretación de los resultados se realiza en función de la estimación del riesgo:
Bajo riesgo . Existe una probabilidad de más del 99% de que el bebé no presente ninguna de las anomalías cromosómicas analizadas. Sin embargo, no puede descartar por completo la posibilidad de afectación cromosómica fetal.
Alto riesgo . la probabilidad de alteración cromosómica o de microdeleción es elevada. En esta situación el ginecólogo recomendará la realización de una prueba de diagnóstico prenatal invasiva para confirmar el diagnóstico.
El resultado viene expresado en porcentaje de riesgo, que oscila entre 0,01 y 99%.
En definitiva, a pesar de su alta fiabilidad, se trata de un test de cribado, por lo que, en caso de resultado positivo, este debe ser confirmado mediante una prueba invasiva (amniocentesis o biopsia corial).
¿Qué es capaz de detectar el test prenatal?
Trisomías. En lugar de dos cromosomas, uno de la madre y otro del padre, hay un tercero. Dependiendo del cromosoma que se trate, puede ser:
Trisomía 21. Síndrome de Down.
Trisomía 18. Síndrome de Edwards.
Trisomía 13. Síndrome de Patau.
Anomalías en los cromosomas sexuales:
Monosomía X (un solo cromosoma X). Síndrome de Turner.
Múltiples cromosomas Y. Síndrome de Klinefelter.
Están ausentes pequeños fragmentos cromosómicos.
Está ausente un fragmento del cromosoma 22 (22q11.2). Síndrome de DiGeorge.
Otras microdeleciones que dan lugar al síndrome de deleción 1p36, el síndrome de Cri-du-chat, el síndrome de Angelman y el síndrome de Prader-Willi.
Triploidía.
Sexo fetal.
¿Cuánto tardan los resultados de un test no invasivo?
Los resultados de los test no invasivos están basados en el análisis del ADN fetal presente en la sangre materna. Estos pueden estar disponibles, aproximadamente, en una semana.
¿Qué fiabilidad tienen los test prenatales no invasivos?
La fiabilidad del test prenatal no invasivo es muy elevada, situándose por encima del 99% respecto a la detección de las trisomías más habituales (síndromes de Down, Patau y Edwards y las que afectan a los cromosomas sexuales).
La tasa de falsos positivos se sitúa en menos del 0,1%. De igual modo, la tasa de falsos negativos en el test prenatal es muy baja y se relaciona con niveles de ADN fetal muy bajo en la sangre materna.
Limitaciones del test prenatal
Algunas situaciones pueden limitar la utilidad del test, por lo que no se recomienda su uso. Entre ellas se encuentran:
Embarazo inferior a 9 semanas en el momento de la prueba.
Embarazo de 3 o más fetos o con constancia de gemelo evanescente.
Embarazo mediante donación ovular. En este caso el riesgo de que el bebé presente alguna alteración cromosómica suele ser menor, dado que estos óvulos suelen proceder de mujeres jóvenes y correctamente estudiadas. Pero si fuera necesario, se podría realizar con la misma eficacia diagnóstica que en fetos procedentes de óvulos propios de la madre.
Haber recibido recientemente una transfusión sanguínea, trasplante de médula ósea o terapia de células madre.
Los mosaicismos fetales de las trisomías, así como alteraciones parciales de los cromosomas estudiados, pueden no ser detectados.
A pesar de su alta fiabilidad, se trata de un test de cribado, por lo que, en caso de resultado positivo, éste debe ser confirmado mediante una prueba invasiva (amniocentesis o biopsia corial). En cualquier caso, la realización del test prenatal está exenta de riesgo para el feto y para la madre.
Artículo especializado
Microdeleción: ¿Qué es y qué síndromes genera?
La ausencia de una parte de material genético en los cromosomas puede determinar la aparición de diversos síndromes congénitos.
¿Qué son las microdeleciones?
Las microdeleciones son anomalías cromosómicas causadas por pequeñas pérdidas de material genético que aparecen de forma congénita y que están presentes durante la gestación. Estas ausencias de genes determinan la aparición de graves síndromes que determinan problemas del desarrollo en el recién nacido.
Los diferentes síndromes dependen del cromosoma y la zona específica en la que se ha producido la pérdida genética. Estos tienen características clínicas distintivas que permiten muchas veces sospechar la microdeleción subyacente.
Cromosomas e información genética
La información genética presente en el ADN se organiza dentro de cada célula en los cromosomas. Los seres humanos tenemos 23 pares de cromosomas (dos copias de cada par, es decir 46). Los primeros 22 pares se numeran del 1 al 22. El último par determina el sexo. Las niñas tienen dos cromosomas X y los niños un cromosoma X y un cromosoma Y. Los problemas de salud y de desarrollo aparecen cuando falta (microdeleciones) o sobra algún cromosoma o una parte de ellos.
La mayoría de las deleciones cromosómicas ocurren como un acontecimiento al azar durante la formación de las células reproductoras (óvulos o espermatozoides) o durante el desarrollo fetal temprano. De este modo, no existen fatores hereditarios y los padres presentan cromosomas normales. En estas microdeleciones la edad materna tampoco influye.
Las anomalías cromosómicas, incluyendo las microdeleciones más comunes, pueden ser detectadas gracias a los test prenatales no invasivos , sin riesgo para la madre ni el bebé.
¿Cuáles son los síndromes de microdeleción?
Existen diferentes síndromes causados por microdeleción con características clínicas diferenciadas. Entre los más comunes se encuentran:
Síndrome de Smith-Magenis
El Síndrome de Smith-Magenis tiene una incidencia cercana a 1 caso por cada 25.000 recién nacidos.
Corresponde a una deleción en el cromosoma 17, en concreto la banda p11.2 (17p11.2).
Se caracteriza por múltiples anomalías congénitas, junto con retraso mental. El fenotipo característico incluye los siguientes signos:
Braquicefalia o aplanamiento de la parte posterior de la cabeza.
Hipoplasia mediofacial, con desarrollo insuficiente de los huesos maxilares.
Mandíbula prominente.
Voz ronca.
Alteraciones del sueño.
Síndrome de Prader-Willi
El Síndrome de Prader-Willi afecta a 1 de cada 15.000-30.000 recién nacidos.
Está causado por la ausencia de contribución paterna de una región en el cromosoma 15 (en concreto la 15q11.2q11.3).
Se caracteriza por la presencia de:
Déficit cognitivo y trastorno de conducta.
Hipotonía severa.
Dificultad para alimentarse durante la lactancia.
Hiperfagia entre 1 y 6 años de edad con desarrollo gradual de obesidad mórbida.
Hipogonadismo en ambos sexos.
Talla baja.
Síndrome de Angelman
Este síndrome tiene una incidencia 1 caso entre cada 10.000-20.000 nacimientos.
Está causado por la ausencia de la contribución materna de la región del cromosoma 15 (en concreto el gen UBE3A).
Se caracteriza, en más de 90% de los pacientes, por una historia prenatal y de recién nacido normal con un perfil metabólico normal y un sistema nervioso central estructuralmente normal, pero con la posterior presencia de:
Retraso mental severo.
Ausencia de lenguaje.
Ataxia, temblor de las extremidades y convulsiones.
Microcefalia.
Estrabismo.
Hipopigmentación de piel y ojos.
Protrusión lingual con alteración de la succión-deglución y problemas de alimentación.
Boca amplia con dientes separados y mandíbula prominente.
Brazos flectados en la marcha.
Hipersensibilidad al calor.
Atracción por el agua.
Alteraciones del sueño.
Síndrome de DiGeorge
El síndrome de DiGeorge es el síndrome por microdeleción más común. Presenta una incidencia de uno de cada 2.000-4.000 recién nacidos vivos.
Se trata de un cuadro con expresión variable, causado por una deleción submicroscópica en el cromosoma 22 (22q11.2) en el 95 % de los casos. Las personas afectadas también presentan un mayor riesgo de autismo, trastorno de déficit de atención y otras afecciones sociales y neurológicas.
Cursa con:
Cardiopatía.
Anomalías del paladar.
Hipocalcemia.
Inmunodeficiencia.
Trastorno del aprendizaje.
Facies característica.
Síndrome de Cri-Du-Chat
El síndrome de “cri du chat” o “maullido de gato” es una enfermedad hereditaria rara que afecta a 1 de cada 20.000-50.000 recién nacidos.
Está causado por la deleción de material genético en uno de los brazos del cromosoma 5.
Se caracteriza por el hecho de que el bebé lactante afectado presenta un llanto similar al maullido de un gato.
Se caracteriza, además, por la presencia de los siguientes signos clínicos:
Anomalías cardíacas
Discapacidad intelectual.
Bajo peso al nacer y crecimiento lento.
Retraso de las habilidades motoras.
Pérdida de la audición.
Ojos separados e inclinados hacia abajo.
Aparición de un pliegue adicional de piel sobre el ángulo interior del ojo.
Orejas de implantación baja o de forma anormal.
Fusión o formación parcial de membranas en los dedos de las manos o los pies.
Escoliosis.
Una sola línea en la palma de la mano.
Microcefalia.
Síndrome de Deleción 1p36
El síndrome de deleción 1p36 es una anomalía cromosómica que puede afectar a 1 de cada 5.000 nacidos vivos.
El síndrome de deleción 1p36 está causado por la ausencia genética que afecta principalmente al extremo terminal del brazo corto del cromosoma 1.
Los signos distintivos del síndrome incluyen:
Rasgos faciales dismórficos (microbraquicefalia, una fontanela anterior amplia con retraso de cierre, cejas rectas, ojos hundidos, puente nasal ancho y deprimido, protrusión del tercio medio facial, orejas anómalas de implantación baja y rotadas posteriormente, surco subnasal largo y barbilla puntiaguda).
Dedos y pies cortos.
Hipotonía.
Retraso del desarrollo.
Discapacidad intelectual.
Crisis epilépticas.
Defectos cardíacos.
Alteración o ausencia del habla.
Restricción del crecimiento de inicio prenatal.
Anomalías esqueléticas.
Anomalías en los genitales.
Síndrome de Wolf-Hirschhorn
El síndrome de Wolf-Hirschhorn es un trastorno genético que afecta a 1 de cada 20.000-50.000 recién nacidos. Es más frecuente en mujeres.
Está causado por la deleción de material genético cerca del extremo del brazo corto del cromosoma 4. El tamaño de la deleción varía, mostrándose síntomas más graves en las deleciones mayores.
Se caracteriza por la presencia de los siguientes signos:
Apariencia facial característica (microcefalia, nariz ancha, frente alta con glabela prominente, ojos separados, cejas arqueadas y altas, surco nasolabial corto, boca curvada hacia abajo, orejas malformadas y mandíbula muy pequeña).
Retraso del crecimiento y del desarrollo.
Discapacidad intelectual.
Bajo tono muscular (hipotonía) y convulsiones.
Anomalías de los huesos.
Defectos congénitos cardíacos.
Pérdida de la audición.
Malformaciones del tracto urinario.
Anormalidades estructurales del cerebro.
Síndrome de Jacobsen
El síndrome de Jacobsen es una cromosomopatía infrecuente que afecta a uno de cada 100.000 nacimientos.
Está causado por una deleción terminal del brazo largo del cromosoma 11.
Los principales rasgos clínicos incluyen:
Retraso psicomotor y del crecimiento.
Trigonocefalia (forma triangular o en cuña de la frente).
Dismorfia facial característica.
Hematomas y petequias generalizadas.
Pies zambos.
Anomalías digestivas y renales.
Síndrome de Langer-Giedion
El síndrome de Langer-Giedion es un síndrome muy poco frecuente que afecta a menos de un caso por cada millón de nacimientos.
Está causado por una deleción del cromosoma 8 (8q 23.3-q24.11) que afecta a los genes EXT 1 y TRPS 1.
El espectro clínico incluye:
Retraso mental.
Talla baja.
Microcefalia.
Dismorfismo facial.
Exóstosis o formaciones de tejido óseo en el conducto auditivo.
Anormalidades esqueléticas.
Microdeleciones del cromosoma Y e infertilidad masculina
La infertilidad masculina puede tener su origen en causas genéticas. De este modo, las microdeleciones del cromosoma Y (cromosoma sexual masculino) pueden suponer una causa de infertilidad en el hombre.
El cromosoma Y posee una región denominada AZF o factor de azoospermia. Las deleciones en esta región cromosómica se pueden traducir en trastornos en la producción de esperma. Estos pueden incluir azoospermia o imposibilidad total de producir espermatozoides y oligospermia, en la que la producción de espermatozoides está disminuida.
¿Cuál es el síndrome de microdeleción más común?
El síndrome por microdeleción más frecuente es el síndrome de DiGeorge. En él está ausente un pequeño fragmento del cromosoma 22 (22q11.2), lo que acarrea múltiples alteraciones fetales como defectos cardíacos, anomalías en el paladar y la cara, retraso en el desarrollo e inmunodeficiencia, con infecciones frecuentes.
El asesoramiento genético ofrecido a través de los resultados de los test prenatales no invasivos ayuda a diagnosticar estas microdeleciones, favoreciendo el diagnóstico precoz y ofreciendo las opciones para su tratamiento y manejo oportunos.
Artículo especializado
Grupo sanguíneo en el embarazo: ¿Cuál trae problemas?
Todas las personas tienen un grupo sanguíneo (O, A, B o AB) y un factor Rh (positivo o negativo). El grupo sanguíneo y el factor Rh significan simplemente que la sangre de una persona tiene ciertas características específicas. El grupo sanguíneo se traduce en forma de proteínas en los glóbulos rojos y en los líquidos corporales.
Por otro lado, el factor Rh se identifica con una proteína que recubre los glóbulos rojos. Si esta proteína está presente, la persona tiene un factor Rh positivo. En cambio, si la proteína está ausente, la persona tiene factor Rh negativo.
Los factores Rh se determinan genéticamente. Un bebé puede tener el grupo sanguíneo y el factor Rh de cualquiera de sus padres o una combinación de ambos. Los factores Rh siguen un patrón común de herencia genética. El gen Rh positivo es dominante (más fuerte) e incluso cuando se combina con un gen Rh negativo, el positivo prevalece.
El problema tiene lugar cuando se ponen en contacto dos grupos sanguíneos con distintos factores Rh, provocando una respuesta inmunitaria. Esto puede suceder en una transfusión sanguínea, o cuando la madre embarazada es factor Rh positivo y el embrión en gestación es Rh negativo.
Estas manifestaciones se traducirán en una serie de eventos clínicos de distinta gravedad, según la cantidad sanguínea expuesta al contacto.
Grupo sanguíneo: sistema AB0
El grupo sanguíneo se basa en la presencia o ausencia de ciertos antígenos que se encuentran en la superficie de los glóbulos rojos. En este sentido, también son relevantes los anticuerpos que circulan en la sangre que reconocen estas células como propias.
Los glóbulos rojos pueden clasificarse en tres tipos: A, B y 0, de acuerdo con la presencia o ausencia de antígenos en la superficie.
Grupo A . Los glóbulos rojos de los individuos del grupo A tienen antígenos del tipo A en su superficie y anticuerpos anti-B en la sangre. Se estima que este grupo puede alcanzar a un tercio de la población.
Grupo B . Los glóbulos rojos presentan en su superficie antígenos tipo B y anticuerpos anti-A en la sangre. Cerca del 20% de la población es del grupo B.
Grupo AB . Los antígenos que aparecen en la superficie de los glóbulos rojos son de los dos tipos: A y B y no presentan anticuerpos en el plasma sanguíneo. Es el grupo sanguíneo menos común, alcanzando tan solo al 5% de la población.
Grupo 0. Los glóbulos rojos no presentan ningún antígeno en su superficie, por lo que tienen anticuerpos anti-A y anti-B en el plasma sanguíneo. Es el grupo mayoritario, con más del 40 % de la población.
¿Qué tipo de sangre tiene problemas en el embarazo?
Como se ha dicho, cuando un glóbulo rojo de un grupo sanguíneo entra en contacto con la sangre de otro grupo (transfusión o embarazo) provoca una reacción inmunológica en el huésped. En el caso de que exista una diferencia de grupo sanguíneo (A, B, AB y 0) entre la madre y el feto en gestación, no suelen aparecer problemas de consideración. De este modo, no hay afectación prenatal y la postnatal es leve o moderada.
Esta situación es debida a que los anticuerpos del sistema AB0 no son capaces de atravesar la placenta o lo hacen en cantidades muy limitadas, ya que se tratan del tipo IgM.
La incompatibilidad del factor Rh es la situación que tiene mayor importancia, ya que los anticuerpos anti-D son de clase IgG, capaces de atravesar la placenta, pudiendo provocar la enfermedad hemolítica del recién nacido. En esta los glóbulos rojos del feto son atacados por los anticuerpos maternos, ya que los reconoce como extraños y se pone en marcha un mecanismo para su destrucción.
Esta situación sucede cuando una mujer Rh negativa se queda embarazada, siendo el bebé Rh positivo. En este caso, se puede producir un intercambio entre la sangre del bebé y la de la madre durante el embarazo y, especialmente, durante el parto.
Cuando se trata del primer embarazo esta situación no suele ser peligrosa. Al contrario de lo que ocurre con el sistema AB0, los anticuerpos anti-Rh no se encuentran en el plasma de los individuos Rh negativo hasta que las células del sistema circulatorio de estos individuos hayan estado en contacto con el antígeno Rh positivo con anterioridad.
De este modo, tras ese primer embarazo, la madre tendrá anticuerpos frente al antígeno Rh positivo. En un segundo embarazo con las mismas circunstancias se produciría el ataque de los anticuerpos de la madre frente a los antígenos de los glóbulos rojos del bebé, provocando su hemólisis.
Enfermedad hemolítica del recién nacido
La enfermedad hemolítica del recién nacido es el proceso en el que se produce la destrucción acelerada de los glóbulos rojos fetales por parte de los anticuerpos maternos, generando una anemia grave en el bebé.
El intento de generación de nuevos hematíes para suplir esa carencia determina la presencia de eritroblastos, glóbulos rojos inmaduros incapaces de cumplir su función de forma correcta.
La destrucción masiva de hematíes produce una sustancia de deshecho, la bilirrubina, que a partir de ciertos niveles es tóxica. En primera instancia provoca ictericia, pero en casos graves, cuando la concentración va en aumento y se acumula, puede provocar trastornos neurológicos e incluso la muerte.
¿Qué pasa cuando una mujer embarazada es Rh negativo?
Como se ha dicho, cuando la madre es Rh negativo y el feto Rh positivo, esta puede generar anticuerpos frente al Rh de su hijo.
Esta reacción puede llegar a eliminar los glóbulos rojos del bebé, provocando anemia en el momento del parto y también a lo largo del embarazo.
Para evitar esta situación, en madres con Rh negativo se lleva a cabo el test de Coombs indirecto, una prueba que detecta anticuerpos frente al Rh positivo del feto capaces de atravesar la placenta, causando la enfermedad hemolítica del recién nacido.
En esos casos, se administra a la madre de forma preventiva en torno a las 28 semanas de gestación una gammaglobulina anti-D que impide la producción de anticuerpos contra las células sanguíneas del bebé.
Tras el parto, si se confirma que el Rh del recién nacido es positivo, durante las primeras 72 horas, habrá que repetir la dosis de gammaglobulina a la madre.
Después del embarazo, se pueden realizar transfusiones si el bebé padece de una anemia grave y administrar líquidos intravenosos si la tensión arterial es baja. En los casos en los que el bebé tiene ictericia, existe un procedimiento llamado exanguinotransfusión en el que se extrae sangre del bebé con un nivel alto de bilirrubina y se reemplaza por sangre nueva con un nivel normal. Además de bajar el nivel de bilirrubina, se aumenta de esta forma el recuento de glóbulos rojos. En los casos más leves de bilirrubinemia, está indicada la fototerapia para ayudar al bebé a eliminar el exceso de bilirrubina. También existe la opción de administrar Inmunoglobulina intravenosa para ayudar al sistema inmune del bebé y reducir la hemólisis de los glóbulos rojos.

Artículo especializado
Síndrome de Smith-Magenis: ¿Qué es y cómo detectarlo?
Las microdeleciones son anomalías cromosómicas causadas por pequeñas pérdidas de material genético que pueden determinar la aparición de síndromes congénitos que cursan con problemas en el desarrollo del recién nacido.
Los diferentes síndromes dependen del cromosoma y la zona específica en la que se ha producido la pérdida genética. Uno de ellos es el Síndrome de Smith-Magenis.
¿Qué es el síndrome de Smith-Magenis?
El síndrome de Smith-Magenis está causado por una deleción o pérdida de material genético en el cromosoma 17 (banda G11.2p). Recientemente se ha comprobado que la pérdida o las mutaciones del gen RA/1 situado en esa región cromosómica podría ser el causante específico del síndrome.
El síndrome queda definido por un conjunto de características físicas y de conducta que incluyen retraso mental.
Se estima que el síndrome de Smith-Magenis afecta a uno de cada 25.000 recién nacidos (niños y niñas), si bien la incidencia podría ser mayor.
En la mayoría de los casos, la deleción de material genético tiene lugar de forma accidental, por lo que se puede calificar como un síndrome congénito, pero no hereditario.
Cromosomas e información genética
La información genética presente en el ADN se organiza dentro de cada célula en los cromosomas. Los seres humanos tenemos 23 pares de cromosomas (dos copias de cada par, es decir 46). Los primeros 22 pares se numeran del 1 al 22. El último par determina el sexo. Las niñas tienen dos cromosomas X y los niños un cromosoma X y un cromosoma Y. Los problemas de salud y de desarrollo aparecen cuando falta (microdeleciones) o sobra algún cromosoma o una parte de ellos. En el caso del síndrome de Smith-Magenis, el cromosoma afectado es el 17.
La mayoría de las deleciones cromosómicas ocurren como un acontecimiento al azar durante la formación de las células reproductoras (óvulos o espermatozoides) o durante el desarrollo fetal temprano. De este modo, no existen fatores hereditarios y los padres suelen presentar cromosomas normales. En estas microdeleciones la edad materna tampoco influye.
Las anomalías cromosómicas, incluyendo las microdeleciones más comunes, pueden ser detectadas gracias a los test prenatales no invasivos , sin riesgo para la madre ni el bebé.
Síndrome de Smith-Magenis: caracteristicas
Los afectados por el síndrome de Smith-Magenis presentan una gran variedad de signos y síntomas que se hacen más evidentes con la edad.
El nacimiento del bebé suele ser a término y con unas características normales. Posteriormente empiezan a aparecer hipotonía, letargia y problemas en la alimentación, lo que determina una ralentización del crecimiento.
El fenotipo característico del síndrome de Smith-Magenis incluye los siguientes signos:
Características físicas y fisiológicas
Braquicefalia o aplanamiento de la parte posterior de la cabeza.
Hipoplasia mediofacial, con desarrollo insuficiente de los huesos maxilares.
Mandíbula prominente.
Voz ronca.
Manos y pies cortos y anchos.
Talla baja.
Escoliosis.
Anomalías en el oído medio.
Inestabilidad al caminar.
Piel áspera.
Hipotonía.
Anomalías renales o urinarias.
Alteraciones cardíacas.
Trastornos tiroideos.
Desprendimiento de retina.
Estreñimiento.
Características cognitivas y de comportamiento
Retraso mental.
Alteraciones del sueño.
Retraso del lenguaje.
Problemas para aprender a leer.
Estereotipias o movimientos repetitivos y convulsiones.
Conductas autoagresivas.
Diagnóstico
Con un test prenatal no invasivo se puede detectar la mayoría de las deleciones 17p11.2, sin ningún riesgo para la madre ni el bebé, a partir del material genético del feto presente en la sangre materna. Otras alternativas son las pruebas prenatales invasivas como la amniocentesis o la biopsia corial.
Con posterioridad al nacimiento, el diagnóstico precoz del síndrome es de gran ayuda a los padres para aprender las características de la enfermedad y las necesidades específicas de los niños afectados.
Este síndrome por microdeleción comparte características y signos con otros síndromes y patologías, de ahí las dificultades para establecer un diagnóstico clínico. Entre ellos se cuentan el síndrome de Prader-Willi (deleción en el cromosoma 15), el síndrome de Williams (deleción en el cromosoma 7), los trastornos del espectro autista y el trastorno por déficit de atención e hiperactividad (TDAH).
Tratamiento
El síndrome de Smith-Magenis, como el resto de los síndromes cromosómicos congénitos no se puede curar. Sin embargo, muchos de los signos y características del síndrome pueden mejorar con diferentes tratamientos establecidos por un equipo médico multidisciplinar. Algunos de estos tratamientos incluyen, entre otros:
Medicación psicotrópica para estabilizar los cambios del humor, disminuir la ansiedad y aumentar la atención.
Programas de intervención en la primera infancia, con educación especial individualizada y seguimiento en el resto del periodo educativo.
Terapia del habla y del lenguaje.
Modificación e intervención del medio, mejora y apoyo de las interacciones sociales.
Plan integral de apoyo conductual para el hogar y la escuela.
Terapias conductuales para ayudar a minimizar las conductas agresivas en el entorno escolar.
Manejo terapéutico del trastorno del sueño.
¿Cuál es la enfermedad que se da en el cromosoma 17?
Las pérdidas de material genético en zonas específicas del cromosoma 17 (parte del brazo corto del cromosoma 17, el segmento 17p11.2) dan lugar al síndrome de Smith-Magenis, llamado así por los apellidos de sus dos descubridoras.
El asesoramiento genético ofrecido a través de los resultados de los test prenatales no invasivos ayuda a diagnosticar las microdeleciones, como la producida en el síndrome de Smith-Magenis, favoreciendo el diagnóstico precoz y ofreciendo las opciones para su tratamiento y manejo oportunos.

Artículo especializado
Hematoma en la placenta: síntomas y tratamiento
Los hematomas placentarios son acúmulos de sangre que se producen por una hemorragia intrauterina en el transcurso del embarazo. Pueden dar lugar al desprendimiento de la placenta y otras complicaciones graves.
Hematomas en el embarazo
Los hematomas aparecen al desarrollarse acúmulos de sangre dentro de la cavidad endometrial, situada entre la capa más superficial del útero y el tejido corial.
No todos los hematomas intrauterinos son peligrosos. Sin embargo, la aparición de un hematoma hace que el embarazo pasa a considerarse de riesgo y se tiene una mayor precaución y control para poder valorar su evolución.
El pronóstico del hematoma depende de sus dimensiones, ubicación y el momento del diagnóstico en el embarazo (no es lo mismo un hematoma en el primer trimestre de la gestación que en el último). De este modo, un hematoma pequeño suele tener un buen pronóstico, con un escaso riesgo de aborto al inicio del embarazo. Por el contrario, un hematoma de gran tamaño ubicado en una zona peligrosa, acompañado de dolor abdominal y un sangrado abundante, en el último trimestre puede ir asociado a un riesgo elevado de desprendimiento prematuro de la placenta normoinserta o abruptio placentae . Este se define por la separación aguda parcial o completa de la placenta respecto a la pared uterina, que sucede después de la semana 20, provocando un 20 % de mortalidad fetal y hasta un 1 % de mortalidad materna.
Por otro lado, según la localización del hematoma, este se puede clasificar en hematoma subamniótico, subcoriónico y retroplacentario, de acuerdo a su relación entre la cavidad uterina y el lecho placentario .
En función de las circunstancias específicas de cada caso el médico tendrá que valorar la mejor solución.
¿Qué es un hematoma retroplacentario?
El hematoma retroplacentario (HRP) se define por el desprendimiento prematuro de la placenta normalmente insertada, de forma parcial o total.
Se trata de un sangrado de alta presión, que se produce como consecuencia de la rotura de las arterias espirales de la placenta, a nivel de la decidua basal materna o capa que recubre el útero.
El hematoma puede interrumpir en mayor o menor grado los intercambios materno-fetales y producir trastornos hemodinámicos, anomalías de la coagulación y sufrimiento fetal agudo de intensidad variable.
Se trata de una de las causas principales de hemorragia en la segunda mitad del embarazo, constituyendo una urgencia obstétrica que puede llegar a afectar hasta el 1 % de los embarazos.
En muchas ocasiones se puede sospechar la presencia de un hematoma retroplacentario debido a los signos que preceden al parto (hemorragias, anomalías del ritmo cardíaco fetal, dolor) y el diagnóstico se confirma durante el alumbramiento por la presencia de coágulos entre la cara placentaria materna y la pared uterina. Con menor frecuencia, el desprendimiento placentario es asintomático y se descubre después del parto.
La visualización ecográfica del hematoma puede servir para confirmar la sospecha diagnóstica. Sin embargo, el desprendimiento no suele tener traducción ecográfica. De igual manera, el estudio histológico de la placenta no siempre confirma el diagnóstico de desprendimiento placentario .
Causas de un hematoma retroplacentario
La formación de un HRP puede obedecer a la ruptura de una arteria decidual materna o la necrosis de una vena decidual. En todos los casos, el origen del desprendimiento de la placenta es la formación del hematoma.
Por otro lado, existen numerosos factores de riesgo de la madre para la formación de un hematoma retroplacentario, entre ellos:
Edad materna superior a 35 años o inferior a 20.
Raza negra.
Madre sola.
Tabaquismo.
Alcoholismo y consumo de drogas.
Hipertensión arterial crónica.
Hiperhomocisteinemia.
Trombofilia.
Diabetes preexistente.
Hipotiroidismo.
Anemia.
Anomalías uterinas.
Preeclampsia.
Traumatismo durante la gestación.
¿Cómo eliminar un hematoma retroplacentario?
El hematoma retroplacentario es una urgencia vital para la madre y el feto. El tratamiento dependerá de las circunstancias del diagnóstico, de la edad gestacional y del estado materno y fetal. En todos los casos, pretenderá equilibrar el estado de la madre y proceder a la evacuación del feto, en la mayoría de los casos por medio de cesárea.
Cuando se produce el desprendimiento prematuro de la placenta a término o cerca del término, el parto debe producirse con celeridad. Si el diagnóstico se sospecha antes del trabajo de parto, en general el nacimiento se debe producir por cesárea.
En cualquier caso, se trata de una urgencia obstétrica que puede requerir la finalización inmediata de la gestación. El grado de urgencia y la vía de finalización de la gestación dependerán del estado materno y fetal.
Por otro lado, cuando el hematoma retroplacentario tiene lugar en el primer trimestre suelen presentar un pronóstico favorable en más de la mitad de los casos, siendo fundamental su detección precoz.
¿Qué pasa si estoy embarazada y tengo un hematoma en la placenta?
En función de la posición y el tamaño del hematoma retroplacentario se puede producir un aborto por efecto mecánico o ciertas complicaciones gestacionales, incluyendo el desprendimiento de la placenta, un parto prematuro y el retraso en el crecimiento fetal.
Los síntomas que pueden aparecer son muy variables. Existe una forma totalmente asintomática en la que el coágulo o coágulos se descubren al examinarla placenta después del alumbramiento. Otros se manifiestan con clínica de parto pretérmino idiopático o metrorragia sin dolor. No obstante, el cuadro clínico del hematoma retroplacentario es el que acompaña al desprendimiento de placenta, con metrorragia, hipertono, la pared abdominal se hace rígida en un estado de contracción permanente y el registro cardiotocográfico fetal es patológico.
En la forma típica, la paciente siente un dolor intenso de aparición brusca. Este dolor persistente se asocia a una contractura permanente en el útero y a contracciones uterinas. La exploración en ocasiones permite visualizar pérdidas sanguíneas anómalas típicamente de aparición tardía, de poca cantidad y negruzcas. El tacto vaginal revela un cuello cervical duro.
En otros casos, existe la ausencia de sangrado uterino, presentándose como una amenaza de parto prematuro y anomalías del ritmo cardiaco fetal que se relacionan con un sufrimiento fetal agudo.
La mortalidad y la morbilidad perinatales se relacionan con la prematuridad, el CIR (crecimiento intrauterino restringido) y el sufrimiento fetal. También dependen de la extensión del desprendimiento placentario.
Artículo especializado
Ecografía de las 20 semanas: ¿Qué detecta?
La ecografía es una técnica por imagen no invasiva que permite hacer un seguimiento adecuado del desarrollo fetal durante el embarazo. A lo largo de todo el periodo de gestación, es recomendable llevar a cabo al menos tres exploraciones ecográficas.
Una de ellas es la exploración ecográfica del segundo trimestre o ecografía de las 20 semanas. Se trata de una prueba fundamental que permite confirmar el desarrollo normal del bebé y es útil para identificar posibles malformaciones.
La vía abdominal es la preferente y la utilizada clásicamente para la ecografía de las 20 semanas.
¿Que se ve en la eco de las 20 semanas?
La ecografía de las 20 semanas o ecografía morfológica se realiza en todas las embarazadas en el segundo trimestre de embarazo, alrededor de la semana 20 (de la 18 a la 22). A esta edad gestacional ya se ha producido un desarrollo suficiente de los órganos y tejidos fetales, por lo que es posible detectar anomalías mayores.
La ecografía se realiza por medio de un ecógrafo, cuya sonda de ultrasonidos se sitúa sobre el abdomen de la madre. El eco de los ultrasonidos, tras penetrar en los tejidos de diferente densidad, se transforman en imágenes, pudiendo observar los órganos y estructuras fetales para valorar su estado.
De este modo, se obtiene la máxima información sobre el desarrollo fetal, identificando posibles malformaciones.
La prueba suele estar protocolizada e incluye una serie de mediciones y estudios anatómico-morfológicos:
Confirmación del latido cardíaco del feto, incluyendo la visualización del corazón y su estructura anatómica.
Valoración de la cantidad de líquido amniótico y la placenta.
Estudio pormenorizado de la anatomía fetal. Este incluye diferentes biometrías cefálicas (tamaño del cráneo y diferentes partes de la cabeza). También se estudia el cuello, la columna, el tórax (pulmones y diafragma), abdomen (estómago, vesícula biliar, intestinos, riñones y vejiga) y las extremidades para descartar cualquier malformación o alteración morfológica.
Evaluación del crecimiento fetal, teniendo en cuenta la edad gestacional estimada en el primer trimestre.
Análisis de la estructura uterina y las zonas anatómicas próximas.
Estudio de los genitales, por lo que se puede averiguar el sexo del bebé.
La duración del estudio es de unos 20-30 minutos en su totalidad.
¿Cuál es la ecografía más importante en el embarazo?
Durante toda la gestación se recomienda llevar a cabo al menos 3 estudios ecográficos. Todos ellos tienen su relevancia y razón de ser.
La primera de ellas, en torno a la semana 12 de gestación, evalúa, principalmente, el riesgo de cromosomopatías y síndromes genéticos. En la tercera, entre las semanas 34 y 36 del embarazo, se evalúa, principalmente el crecimiento y desarrollo fetal.
Finalmente, la ecografía de la semana 20 es señalada por la Organización Mundial de la Salud como necesaria para calcular la edad gestacional y mejorar la detección de anomalías fetales y embarazos múltiples. Además, esta permite a los padres una mayor capacidad de decisión en relación con las diferentes opciones planteadas a partir de la eventual detección de alguna anomalía.
¿Cómo está un bebé en la semana 20?
En la semana 20 de gestación el feto ya es lo suficientemente grande como para poder observar sus órganos con claridad. El grado de desarrollo de los órganos y tejidos permite la detección de anomalías mayores.
Órganos como el corazón, los riñones, el hígado, el intestino, los genitales o la columna vertebral ya tienen un grado de desarrollo importante. Tanto la forma como sus proporciones son parecidas a las que presentará en el momento del nacimiento.
En este momento de la gestación ya se pueden estudiar las cavidades del corazón (aurículas y ventrículos), además de la estructura de los riñones, el hígado, el estómago y otros órganos. También se pueden contar las vértebras de la columna y los dedos de manos y pies (si falta alguno puede ser el signo de otros problemas más graves).
Además, se puede analizar que no existan fisuras en el paladar, la presencia de cristalino en los ojos y la existencia de los huesos largos en las extremidades, así como verificar la alineación de los huesos largos y extremidades inferiores con los pies.
En la epidermis, la capa más superficial de la piel, se empiezan a formar los surcos característicos determinados genéticamente de las manos y plantas de los pies.
Por otro lado, en este momento se puede calcular la circunferencia del abdomen y la cabeza para valorar el crecimiento fetal. Con esta y otras mediciones se puede dirimir el tiempo exacto de gestación, junto con la fecha probable de parto.
Ecografía 3D, 4D y 5D
La ecografía obstétrica utilizada para el seguimiento y diagnóstico de los diferentes problemas de desarrollo del feto está basada en una tecnología “2D”, es decir, imágenes en blanco y negro que ofrecen secciones de los tejidos y órganos. Esta técnica permite explorar el feto para analizar su anatomía e identificar malformaciones.
Además de esta tecnología originaria 2D, hace algunos años se desarrolló la tecnología 3D, permitiendo a partir del procesamiento informático de un barrido en dos dimensiones sobre un tejido concreto, formar una visualización de tres dimensiones. A partir de aquí, se pudo avanzar para ver estas imágenes 3D en movimiento en tiempo real, dando lugar a la aparición de la ecografía 4D.
De hecho, se puede realizar una ecografía de las 20 semanas 4D, ya que los aparatos de alta gama empleados para la ecografía morfológica incorporan diferentes programas 3D y 4D para complementar el estudio de las diferentes estructuras de órganos y tejidos.
Si bien el modo 2D es la base de la ecografía del segundo trimestre, en algunos casos, la aplicación de estas nuevas tecnologías puede resultar de utilidad y de especial interés para el estudio del sistema nervioso, corazón, esqueleto y otros tejidos y sistemas.
Finalmente, la ecografía 5D es otra evolución en la que la mejora del tratamiento de la imagen le otorga mayor variedad de tonos y textura, dando a la imagen un aspecto más real. De este modo, la ecografía 5D de las 20 semanas ofrece imágenes más vistosas, pero sin aportar mejores elementos diagnósticos.

Artículo especializado
Mosaicismo genético: ¿Cómo detectarlo?
Frente a las aneuploidías o anormalidades cromosómicas por exceso o defecto de cromosomas que pueden determinar la aparición de diferentes síndromes congénitos, el mosaicismo genético se da cuando el embrión posee una mezcla heterogénea de células cromosómicamente normales y anormales.
¿Qué es el mosaicismo y cómo se produce?
El mosaicismo cromosómico o mosaicismo fetal es una alteración genética en la que un individuo presenta 2 o más grupos de células con distinta composición genética, supuestamente originadas a partir de un mismo cigoto. Esta afección puede alterar cualquier tipo de célula, incluyendo células sanguíneas, óvulos, espermatozoides y células de la piel.
Cromosomas
La información genética presente en el ADN se organiza dentro de cada célula en los cromosomas. Los seres humanos tenemos 23 pares de cromosomas (dos copias de cada par, es decir 46). Los primeros 22 pares se numeran del 1 al 22. El último par determina el sexo. Las niñas tienen dos cromosomas X y los niños un cromosoma X y un cromosoma Y. Los problemas de salud y de desarrollo aparecen cuando falta o sobra algún cromosoma o una parte de ellos. En el caso del mosaicismo, este exceso o defecto cromosómico no se da en todas las células.
Causas del mosaicismo
Las causas del mosaicismo suelen estar relacionadas con accidentes acaecidos durante la división celular del cigoto durante la gestación, en fases muy tempranas del desarrollo fetal y no parece estar asociada a ningún factor paterno ni materno. Las causas pueden ser de 2 tipos:
En la fase de la división celular en la que los pares de cromosomas son separados (anafase), un cromosoma podría retrasarse durante el momento del replicado cromosómico. De este modo, este cromosoma quedaría excluido, dando como resultado después de la división una célula con un cromosoma normal y otra célula con un cromosoma de menos. Si la pérdida de dicho cromosoma no interfiere con la viabilidad y replicación de la célula, ambas seguirán replicándose y el nuevo individuo presentará un mosaicismo de ambos tipos celulares (normales y anormales).
En un segundo tipo de accidente, un cromosoma se replicaría sin dividirse adecuadamente en dos partes en movimiento hacia los polos opuestos de la célula, yendo las dos mitades al mismo polo. El resultado sería tras la división una célula con un cromosoma extra y una célula con un cromosoma de menos.
De este modo, el mosaicismo afecta a la información genética de unas células, pero no de todas. Un individuo que presenta una trisomía en mosaico significa que algunas de sus células tienen tres copias de un cromosoma, mientras que otras tienen dos, es decir, son normales.
Por ejemplo, el mosaicismo cutáneo se daría cuando la afectación de las células de la piel determinara dos o más tipos de células genéticamente distintas que darían diferentes tipos de piel mezcladas. Estos tipos de células producirían diferente cantidad de pigmento, resultando en áreas de piel con distintas tonalidades, por la distinta cantidad de melanina genéticamente programada en las diferentes líneas celulares.
Mosaicismo de línea germinal
El mosaicismo de línea germinal hace referencia al mosaicismo que afecta a los óvulos y al esperma. De este modo, algunos óvulos o espermatozoides tienen una configuración cromosómica normal y otros no. Si uno de los óvulos o espermatozoides afectados participara en un proceso fecundativo, el niño podría presentar una anomalía cromosómica.
No es posible determinar si una persona tiene mosaicismo de línea germinal.
Mosaicismo placentario
El mosaicismo placentario o mosaicismo confinado a la placenta se define como la presencia de dos o más líneas celulares genéticamente diferentes que están circunscritas a la placenta y no están presentes en el feto.
¿Cómo identificar un mosaicismo?
El mosaicismo genético se puede diagnosticar de diferentes maneras. Cuando existe mosaicismo en un feto que se está gestando, puede haber células del feto con diferente composición cromosómica en la sangre de la madre. En este caso, los test prenatales no invasivos a partir de una muestra de sangre materna pueden ser problemáticos y dar como resultado un falso negativo.
En el caso de análisis de sangre fetal, el diagnóstico puede ser más complicado en función de las células afectadas por el cambio cromosómico. Por ejemplo, un análisis de sangre puede servir para diagnosticar mosaicismo si el cambio cromosómico está ubicado en células como las de la piel.
¿Qué diferencia hay entre el síndrome de Down y el mosaico?
En general, las personas con mosaicismo presentan los mismos problemas que las que presentan un síndrome por anomalías cromosómicas sin mosaicismo. De este modo, una persona con trisomía 21p en mosaico posiblemente tenga muchas de las mismas características que las personas con trisomía 21p sin mosaico (síndrome de Down). Sin embargo, debido a que los individuos con mosaicismo tienen menos células con la anomalía genética, muchos de los síntomas asociados al síndrome pueden aparecer de una forma más leve.
Desafortunadamente, no es posible predecir con exactitud de qué modo el mosaicismo puede llegar a afectar a una persona, ya que no se puede saber con exactitud las células que presentan la anomalía genética.

Artículo especializado
Síndrome de Klinefelter: ¿Qué es y cómo detectarlo?
Las aneuploidías son anomalías que aparecen cuando se produce un exceso o defecto en los cromosomas. Las que afectan a los cromosomas sexuales son las más habituales. Entre ellas destaca el síndrome Klinefelter, un trastorno genético en el que aparece un cromosoma X adicional en los hombres. Se trata de un trastorno congénito, pero en ocasiones no se diagnostica hasta la edad adulta.
¿Qué produce el síndrome de Klinefelter?
El síndrome de Klinefelter es la primera anomalía de los cromosomas sexuales que ha sido descrita en humanos y que tiene una incidencia de 1 de cada 1.000 varones nacidos.
Cada célula del cuerpo humano contiene en su información genética 23 pares de cromosomas, es decir, 46. La mitad proviene de la madre y la otra mitad del padre. Los cromosomas contienen el material genético con la información necesaria para el correcto desarrollo y funcionamiento del organismo. Los pares de cromosomas del 1 al 22 se denominan autosomas (ADN autosómico) y el par 23 (XX en las mujeres y XY en los hombres) constituyen los cromosomas sexuales.
El síndrome de Klinefelter no se hereda, sino que ocurre como un evento aleatorio durante la formación de las células reproductivas ( óvulos y espermatozoides) que tiene como resultado la presencia de una copia extra del cromosoma x en cada célula (47,XXY).
El síndrome de Klinefelter solo tiene lugar en varones. En un 75 % de los casos se produce un cariotipo XXY. Aproximadamente en un 20 % se alternan diferentes composiciones cromosómicas (mosaicos cromosómicos), es decir, XY en el cromosoma sexual en unas células y XXY en otras. También existen otras variantes minoritarias que incluyen las combinaciones XXYY, XXXY, y XXXXY en los cromosomas sexuales.
Las alteraciones de los cromosomas sexuales suelen ser fenómenos aislados, en los que el único factor predisponente conocido es la edad materna avanzada.
¿Cómo son los niños con síndrome de Klinefelter?
Las aneuploidías de los cromosomas sexuales se caracterizan por una gran variabilidad clínica entre individuos. En cualquier caso, suele tratarse de individuos altos y delgados, con piernas largas. Físicamente no aparecen signos diferenciales hasta llegar a la pubertad. En ese momento empiezan a observarse signos de hipogonadismo, que impide que el cuerpo produzca esperma y testosterona, con tendencia a la obesidad. Esto significa que con frecuencia en la edad adulta serán estériles en un 70 % los casos.
Las deficiencias y anormalidades se hacen más notables cuando aparecen más de dos cromosomas X. En estos casos el desarrollo sexual es más deficiente y el déficit intelectual más grave.
Los signos distintivos más frecuentes en preadolescentes afectados son unos genitales pequeños y unas piernas largas.
En bebés y niños de corta edad en ocasiones no se aprecian signos o estos son muy leves e inespecíficos. Pueden presentar músculos débiles con un desarrollo motor lento y retraso en el habla.
¿Cómo se puede identificar el síndrome de Klinefelter?
El médico puede sospechar la presencia del síndrome de Klinefelter cuando se dan los siguientes síntomas y signos:
Niño con retraso leve y comportamiento inmaduro. La mayoría presentan problemas de aprendizaje, especialmente dislexia. El lenguaje y la memoria auditiva son deficientes. Además, presentan dificultades para leer y escribir.
Los trastornos del comportamiento son frecuentes, especialmente inmadurez, inseguridad y timidez. Las personas afectadas suelen presentar problemas para relacionarse con individuos de su grupo de edad y de adaptación social. La depresión es frecuente en estos individuos.
Adolescente con testículos pequeños. Los caracteres sexuales secundarios se desarrollan poco. El vello corporal es escaso y la distribución puede ser ginecoide, es decir que se desarrolla en zonas típicas femeninas. El tejido graso subcutáneo también puede adoptar una distribución femenina sobre todo a nivel de las caderas, y pueden presentar ginecomastia o agrandamiento de las mamas.
Adulto con hábito eunucoide (órganos sexuales subdesarrollados con afectación en la producción hormonal testicular), ginecomastia y escaso desarrollo muscular. Debido a esta masa muscular poco desarrollada, el cansancio es un síntoma habitual.
Adulto con infertilidad.
Son comunes la aparición de varices y úlceras en las extremidades inferiores.
Existe un mayor riesgo de desarrollar enfermedades autoinmunes.
Los varones con cromosomas sexuales XXY con ginecomastia tienen mayor riesgo de cáncer de mama.
Si bien muchos de los casos de síndrome de Klinefelter no se identifican hasta la edad adulta, con el desarrollo de las técnicas de análisis genético, en los últimos años muchos casos se diagnostican en la etapa prenatal. De este modo, existen test prenatales no invasivos que pueden detectar las anomalías cromosómicas, incluyendo las relacionadas con los cromosomas sexuales como el síndrome de Klinefelter, analizando el ADN fetal presente a partir de una muestra sanguínea materna.
Tratamiento del síndrome de Klinefelter
No existe un tratamiento que pueda revertir las anomalías cromosómicas como las presentes en le síndrome de Klinefelter. Sin embargo, las personas afectadas pueden desarrollarse y llevar una vida normal. Para ello, será necesaria una intervención precoz que contemple un ambiente positivo en casa y en la escuela con métodos de estudio adaptados. También debe incluir un apoyo cognitivo y psicológico adecuado, junto con un seguimiento médico que incluya el tratamiento hormonal.
La terapia hormonal debe incluir el tratamiento sustitutivo con testosterona desde que se inicia la pubertad. Este será necesario para promover el desarrollo de los caracteres sexuales secundarios masculinos, el crecimiento testicular y el aumento de la masa muscular.
En cuanto a la esterilidad, no existe ningún tratamiento que pueda revertirla, si bien se puede potenciar la fertilidad cuando existe una producción mínima de espermatozoides.
Artículo especializado
Aneuploidías: ¿Qué son y cómo se detectan?
Las aneuploidías son anomalías cromosómicas que pueden determinar la aparición de diferentes síndromes congénitos. Los más habituales se pueden detectar mediante la realización de un test prenatal no invasivo.
¿Qué es aneuploidía y ejemplos?
Las aneuploidías son las alteraciones genéticas en las que el número de cromosomas no son los habituales, tanto por exceso como por defecto. De este modo, existen uno o más cromosomas adicionales o faltan uno o más cromosomas.
Cada célula del cuerpo humano contiene en su información genética 23 pares de cromosomas, es decir, 46 cromosomas. La mitad proviene de la madre y la otra mitad del padre. Los cromosomas contienen el material genético con la información necesaria para el correcto desarrollo y funcionamiento del organismo. Los pares de cromosomas del 1 al 22 se denominan autosomas (ADN autosómico) y el par 23 (XX en las mujeres y XY en los hombres) constituyen los cromosomas sexuales.
Las aneuploidías más comunes son las trisomías. En los nacidos vivos, las aneuploidías más frecuentes son el síndrome de Down (trisomía del cromosoma 21) y el síndrome de Klinefelter (trisomía del cromosoma sexual XXY).
¿Qué provoca las aneuploidías?
La mayoría de estas alteraciones cromosómicas, incluyendo las aneuploidías, se producen durante la producción de los espermatozoides y óvulos. Si un óvulo o espermatozoide defectuoso participa en la fecundación, el embrión se verá afectado por la anomalía. Sin embargo, en la mayoría de los casos se producirá un aborto espontáneo, ya que la mayoría de las aneuploidías son incompatibles con la vida.
¿Cuáles son los tipos de aneuploidías?
Desde un punto de vista teórico, las aneuploidías podrían incluir:
Nulisomías. Cuando faltan los 2 cromosomas homólogos.
Monosomías. Cuando falta un cromosoma.
Disomías. Cuando el número de cromosomas es correcto, pero un par de cromosomas provienen del mismo progenitor.
Trisomías. Cuando en un par de cromosomas se une un tercero.
Tetrasomías y pentasomías. Existen 2 o 3 cromosomas extra, siempre en los cromosomas sexuales.
Las aneuploidías más frecuentes en humanos son las trisomías y monosomías:
Trisomías. En los nacidos vivos las aneuploidías más frecuentes son el síndrome de Down (trisomía del cromosoma 21), seguido por el síndrome de Klinefelter (trisomía XXY).
Monosomías. El más habitual es el síndrome de Turner (monosomía X). No son compatibles con la vida las monosomías de los cromosomas autosómicos.
El resto de las trisomías más comunes, como el síndrome de Edwards (trisomía 18) o el síndrome de Patau (trisomía 13), son incompatibles con la vida y presentan múltiples malformaciones detectables por ecografía. De este modo, el síndrome de Down es la aneuploidía más común causante de discapacidad en la que se enfoca el cribado durante el embarazo.
Aneuploidías autosómicas
Las aneuploidías autosómicas compatibles con la vida dan lugar a síndromes genéticos con importantes consecuencias en el desarrollo del bebé. Se incluyen aquí:
Trisomía 21 (síndrome de Down). Se da en aproximadamente 1 de cada 700 nacimientos. El riesgo aumenta con la edad de la madre. Suele provocar retraso mental y también malformaciones físicas, especialmente defectos cardíacos.
Las trisomías 18 (síndrome de Edwards) y 13 (síndrome de Patau) son más raras y se encuentran en aproximadamente 1 de cada 7.000 nacimientos. El riesgo también se ve aumentado con la edad de la madre. Estas trisomías se asocian a un grave retraso mental y a menudo a graves malformaciones físicas. En la mayoría de los casos no sobreviven al primer año de vida.
Aneuploidías de los cromosomas sexuales
Las aneuploidías de los cromosomas sexuales consisten en la presencia de alteraciones que pueden tener distintas consecuencias, incluyendo malformaciones, trastornos cognitivos y de crecimiento, así como problemas sexuales y de infertilidad.
Síndrome de Klinefelter. Afecta a los hombres y está causado por la presencia de un cromosoma X adicional (XXY). El riesgo aumenta con la edad de la madre. Los afectados suelen presentar una estatura elevada, un ligero retraso mental y problemas de fertilidad.
Síndrome de Jakob. Afecta a los hombres y está causado por la presencia de un cromosoma Y adicional (XYY). Los afectados también presentan una altura mayor de lo normal y pueden presentar retraso en el lenguaje.
Síndrome de Turner. Afecta a las mujeres y está definido por la ausencia de un cromosoma X (X0). Esta monosomía determina baja estatura, infertilidad y trastornos cardiacos y renales.
Síndrome triple X. Afecta a las mujeres y se caracteriza por la presencia de un cromosoma X de más (XXX). En muchas ocasiones no se manifiestan síntomas o estos son leves. Estas mujeres suelen tener una fertilidad normal salvo que se vean afectadas por fallo ovárico precoz o presenten malformaciones genitourinarias que impidan la gestación.
Todavía no ha sido correctamente estudiado si los pacientes tienen más riesgo de que su descendencia, en el caso de que esta sea posible, también presente trisomía (especialmente, triple X o Klinefelter).
Microdeleciones
Las microdeleciones son pequeñas pérdidas de material genético que se asocian con graves problemas del desarrollo en el recién nacido. Incluyen el síndrome de DiGeorge, el síndrome de deleción 1p36, el síndrome de Cri-du-chat, el síndrome de Angelman y el síndrome de Prader-Willi.
El síndrome por microdeleción más frecuente es el de DiGeorge. En él está ausente un pequeño fragmento del cromosoma 22 (22q11.2), lo que acarrea múltiples alteraciones fetales como defectos cardíacos, anomalías en el paladar y la cara, retraso en el desarrollo e inmunodeficiencia, con infecciones frecuentes.
Triploidía
La triploidía es una alteración cromosómica en la que el feto tiene tres copias de cada cromosoma en lugar de dos (69 cromosomas y no 46). Normalmente se produce cuando dos espermatozoides fertilizan un óvulo, pero también puede ocurrir cuando un óvulo diploide es fertilizado por un espermatozoide, o bien en casos en los que un espermatozoide diploide fertiliza un óvulo. Las triploidías no son compatibles con la vida.
Detectar las aneuploidías mediante test prenatales no invasivos
Para el diagnóstico prenatal de las anomalías genéticas más comunes se requiere la obtención de material genético del feto mediante una técnica invasiva como la amniocentesis o la biopsia corial. Se trata de pruebas que llevan asociado un riesgo de pérdida gestacional, por lo que no se recomiendan de forma indiscriminada a todas las mujeres embarazadas. Este tipo de pruebas invasivas están indicadas ante un resultado de alto riesgo en un test prenatal no invasivo o en el cribado combinado de primer trimestre que se realiza de forma generalizada en la semana 12 de gestación.
Sin embargo, gracias a que el ADN del feto circula por el torrente sanguíneo de la madre durante el embarazo y el avance de técnicas genéticas, a partir de una muestra de sangre materna se puede analizar el ADN libre del feto para detectar posibles anomalías cromosómicas.
Los test prenatales no invasivos permiten comprobar el estado del bebé antes de su nacimiento e identificar posibles aneuploidías, así como conocer el sexo fetal.

Artículo especializado
Síndrome DiGeorge: ¿Qué es y cómo detectarlo?
Las anomalías cromosómicas pueden dar lugar a diversos síndromes y problemas de salud. En el caso de la trisomía del cromosoma 21 (presencia de 3 cromosomas en lugar de los 2 preceptivos) el bebé nace con síndrome de Down. De igual modo, la falta de un cromosoma o una parte de él, también se producen complicaciones de salud de origen genético.
Es el caso del síndrome DiGeorge, también conocido como síndrome de deleción del cromosoma 22q11.2, esta pérdida provoca el desarrollo insuficiente de varios sistemas del organismo. Este síndrome abarca varios términos que antes se consideraban afecciones separadas, como el síndrome de Di George, el síndrome velocardiofacial o el síndrome cardiofacial.
¿Qué produce el síndrome de DiGeorge?
En condiciones normales existen dos copias de los 23 cromosomas, cada una heredada de un progenitor. Cuando a una de las copias del cromosoma 22 le falta el segmento 22q11.2 (30-40 genes) esto da lugar a la aparición del síndrome de DiGeorge de manera innata (anomalía del brazo corto del cromosoma 22).
La eliminación de estos genes del cromosoma 22 se produce de forma aleatoria en el espermatozoide o el óvulo de los progenitores, o puede tener lugar en las primeras etapas de la gestación.
La deleción o ausencia de esta parte del cromosoma da lugar a la aparición de trastornos y anormalidades en diferentes órganos y sistemas del organismo.
Se ha calculado que en un 10% de las familias con un caso de síndrome de DiGeorge es por causa hereditaria y se comporta de manera autonómica dominante, es decir, que si existen antecedentes familiares, hay un 50% de posibilidades de transmitirla a los hijos.
¿A qué órganos afecta el síndrome de DiGeorge?
Los defectos congénitos cardíacos y las anomalías en el paladar están presentes en más de un 70% de los casos.
De este modo, los problemas de salud que se asocian al síndrome de DiGeorge pueden ser variables e incluyen anomalías en diferentes órganos y sistemas, incluyendo entre otros:
El corazón.
El sistema inmunitario.
El sistema hormonal.
Aparición de deformidades en el paladar.
Bajos niveles de calcio en la sangre que dan lugar a diferentes complicaciones.
Retrasos en el desarrollo.
Problemas de aprendizaje, emocionales y trastornos de conducta.
Problemas cardíacos
Los defectos cardíacos que provoca el síndrome de deleción del cromosoma 22q11.2 podrían determinar un suministro de oxígeno insuficiente en los tejidos. Este puede venir determinado por diferentes anomalías en la estructura del corazón y de sus vasos sanguíneos. Es el caso de la tetralogía de Fallot, presente en el 92% de los casos.
Trastornos del sistema inmunitario
El timo es la glándula donde maduran las células T, un tipo de leucocitos necesarios para combatir las infecciones. El síndrome de deleción del cromosoma 22q11.2 determina una disfunción del timo que impide que el sistema inmunitario pueda luchar contras las infecciones con normalidad. Además de las infecciones recurrentes durante la infancia, el riesgo de padecer enfermedades autoinmunes durante la edad adulta también se ve aumentado.
Trastornos hormonales
El síndrome de DiGeorge puede determinar un funcionamiento reducido de las glándulas paratiroideas, lo que provoca hipoparatiroidismo. Este trastorno hace que estén presentes niveles reducidos de calcio y niveles elevados de fósforo en la sangre. Esto puede desembocar en dolores musculares, problemas renales, arritmias cardiacas y problemas en el desarrollo físico y mental, entre otras consecuencias.
Anomalías en el paladar y la cara
En el síndrome de DiGeorge también se suelen ver afectados el paladar y la cara, con la aparición de una hendidura en el paladar y la aparición de labio leporino. En estos, el tejido del paladar y del labio superior presentan una deficiencia y no quedan perfectamente unidos, lo que puede ocasionar dificultades en la deglución y en el habla, entre otros problemas.
Además, pueden aparecer ciertos rasgos faciales distintivos. Estos pueden incluir orejas pequeñas de implantación baja, abertura de los ojos reducida, rostro alargado y estrecho y nariz tubular con punta bulbosa, entre otros.
Problemas de desarrollo cerebral y aprendizaje
La deleción del cromosoma 22q11.2 puede causar problemas en el desarrollo mental y el aprendizaje, la conducta y el comportamiento social. Además, existe un mayor riesgo de trastorno por déficit de atención e hiperactividad (TDAH) y trastornos del espectro autista en la etapa infantil. En la edad adulta existe un mayor riesgo de depresión, ansiedad y otros trastornos mentales.
¿Cómo se detecta el síndrome DiGeorge?
Para el diagnóstico prenatal de las anomalías genéticas más comunes, incluyendo la deleción del cromosoma 22q11.2, se requiere la obtención de material genético del feto mediante una técnica invasiva como la amniocentesis o la biopsia corial. Estas pruebas llevan asociado un riesgo de pérdida gestacional, por lo que no se utilizan de forma indiscriminada en todos los embarazos.
En este sentido, el avance en las técnicas genéticas ha permitido desarrollar test prenatales no invasivos que se pueden aplicar como pruebas de cribado a partir de la sangre de la madre. Con ellos es posible identificar los embarazos con un riesgo elevado de presentar una anomalía genética, pero sin poner en riesgo al feto ni a la madre.
Con todas las pruebas que se realizan durante el embarazo , incluyendo las ecografías y controles periódico, además del acceso a test prenatales no invasivos, es muy complicado que una anomalía cromosómica como el síndrome de DiGeorge pase desapercibida.
¿Cuál es la función del cromosoma 22?
Los genes que forman parte del cromosoma 22 están implicados en múltiples funciones y codifican para multitud de órganos y tejidos del organismo. Por lo tanto, sus anomalías y deficiencias pueden tener consecuencias muy diversas, provocando patologías y afecciones en múltiples sistemas del organismo, como las alteraciones cardíacas, el déficit inmunitario y las características dismórficas faciales anteriormente descriptas.
Artículo especializado
CIR en el embarazo: crecimiento intrauterino restringido
El crecimiento fetal durante la gestación está regulado por factores maternos, fetales y placentarios. Todos ellos en coordinación harán que se asegure la llegada de los nutrientes maternos al feto en la forma y proporción adecuadas, consiguiéndose un crecimiento fetal optimo .
El desarrollo normal de estos condicionantes permite al feto obtener el crecimiento que genéticamente está determinado, desarrollando todo su potencial intrauterino. Por el contrario, cuando existen alteraciones en alguno de los factores, se pueden producir retrasos y restricciones en el crecimiento fetal dentro del útero. Es lo que se conoce como crecimiento intrauterino restringido (CIR) .
¿Qué es el CIR o crecimiento intrauterino restringido?
El crecimiento intrauterino restringido (CIR) es una condición en la que el crecimiento del feto dentro del útero se ralentiza o se detiene.
Se trata de un problema relativamente común que puede llegar a afectar a cerca del 10% de los embarazos en diferente grado, manteniéndose como unos de los grandes retos en la práctica obstétrica actual.
La consecuencia principal del CIR es que el bebé no alcance el tamaño previsto, presentando por ello mayor riesgo de padecer complicaciones en el momento de nacer y también tras el parto. Por ejemplo, muerte intrauterina, prematuridad y peor desarrollo neurológico, entre otras.
¿Cuándo se detecta un CIR?
El grado de desarrollo del feto durante el embarazo, así como la posible situación de CIR es detectada a partir de los controles ecográficos periódicos a los que se someten las embarazadas para evaluar el crecimiento fetal.
Cuando el crecimiento fetal durante toda la gestación es inferior a lo esperado, se considera que es un feto con CIR de aparición precoz. En este caso suele deberse a anomalías congénitas o infecciones prenatales.
Por el contrario, cuando el crecimiento es normal y en el tercer trimestre de gestación se ralentiza o se detiene, se tratará de un CIR de aparición tardía, en cuyas causas estarán involucrados con toda probabilidad los factores maternos o placentarios. La causa más común es una insuficiencia placentaria causada por hipertensión materna. El pronóstico en estos casos es bueno.
De este modo, los problemas en el funcionamiento de la placenta suele ser la causa más común. No en vano se trata del tejido encargado de transportar el alimento y el oxígeno al feto.
En cuanto a los factores maternos de riesgo podemos mencionar:
La edad de la madre (menor de 16 o mayor de 35 años).
El bajo nivel socioeconómico.
Haber transcurrido menos de 6 meses entre dos gestaciones.
Consumo de tabaco, alcohol y drogas.
El tratamiento con determinados fármacos por parte de la madre.
Patologías maternas como asma, hipertensión arterial, diabetes mellitus, patologías renales, etc.
Infecciones y parasitosis en la madre (toxoplasmosis, citomegalovirus, malaria, VIH, etc.).
Entre los factores fetales se pueden encontrar:
Anomalías cromosómicas, como el síndrome de Down y otros síndromes genéticos complejos.
Anomalías congénitas con afectación cardíaca, neurológica, etc.
Gestaciones múltiples.
Enfermedades metabólicas.
¿Qué es un CIR tipo 1?
El CIR de tipo 1 tiene lugar durante el primer trimestre del embarazo, es decir, se trata de CIR precoz. En este caso, la disminución en el crecimiento es proporcional, por lo que se produce un menor crecimiento, pero simétrico y armónico en el que todas las medidas ecográficas están disminuidas. Por motivos diversos (incluyendo la genética) el feto recibe menos nutrientes de lo necesario, por lo que su tamaño es más reducido. La insuficiencia placentaria que se produce es severa, la prematuridad mayor y el pronóstico más grave.
Por el contrario, en el CIR tipo 2 o asimétrico se produce a partir de la semana 34 del embarazo, es tardío y es el más frecuente. Generalmente, se asocia a hipertensión materna durante el embarazo. En este caso el crecimiento es desigual, mostrándose una cabeza, los huesos largos y el abdomen más grandes y desproporcionados que el resto del cuerpo. En el tipo 2, los nutrientes se dirigen prioritariamente al cerebro y las partes más importantes del cuerpo, por lo que se desarrollan en mayor medida.
¿Cómo se diagnóstica el crecimiento intrauterino retardado?
Se considera que el peso del recién nacido es bajo cuando es inferior a 2,5 kg, independientemente de si ha sido pretérmino y de su edad gestacional. De igual modo, un bebé pequeño para su edad gestacional es aquel que está por debajo del percentil 10, en las curvas de percentiles, situándose el crecimiento normal en 50, pero sin que concurran trastornos en el desarrollo.
Por el contrario, el crecimiento intrauterino restringido o retardado se produce cuando se ha producido un crecimiento anormal dentro del útero materno. En este caso, el embrión no ha sido capaz de desarrollarse plenamente dentro del vientre materno.
La prueba principal para establecer el diagnóstico de CIR es la ecografía. Las medidas del feto permiten estimar su peso y establecer en qué percentil de crecimiento se encuentra, teniendo en cuenta otros factores como el sexo, origen étnico, etc.
El estudio de crecimiento fetal se complementa con el estudio Doppler, que permite evaluar el funcionamiento de la placenta y el grado de oxigenación del feto.
Cuando se ha establecido el diagnóstico y en función del momento de aparición del retraso de crecimiento, se pueden realizar otras pruebas para valorar las posibles causas:
Indicada en casos graves y antes de la semana 26.
Analítica para el estudio de posibles infecciones y parasitosis.
Analítica para identificar alteraciones compatibles con preeclampsia.
Seguimiento periódico ecográfico del crecimiento fetal.
Para hacer el seguimiento del desarrollo fetal la herramienta más útil es la ecografía obstétrica .
¿Qué pasa cuando el bebé deja de crecer en el vientre?
Los bebés que han experimentado un crecimiento intrauterino restringido presentan un mayor riesgo de sufrir determinados problemas de salud. Además de requerir mayores cuidados y un mayor periodo de hospitalización tras el nacimiento, pueden presentar:
Problemas respiratorios y para alimentarse.
Dificultad para mantener una temperatura estable en el cuerpo.
Recuento sanguíneo anormal.
Hipoglucemia
Menor capacidad inmunitaria.
Problemas neurológicos.
Además, también existirá un mayor riesgo para la salud en etapas posteriores de la vida, incluso en la edad adulta, incluyendo enfermedades cardiovasculares y diabetes, entre otras.
El correcto seguimiento del embarazo permitirá un diagnóstico precoz y oportuno, pudiendo tomar medidas para disminuir las complicaciones presentes y futuras en estos casos.
Artículo especializado
Embarazo anembrionario: síntomas y causas
Existen diversas causas que pueden determinar que un embarazo acabe en aborto en las primeras semanas de gestación. Una de ellas es el embarazo anembrionario.
¿Qué es el embarazo anembrionario?
El embarazo anembrionario es un embarazo en el que se forma un saco gestacional, pero sin un embrión en su interior. Este tipo de embarazo terminará en aborto, debido a que, aunque el huevo fertilizado se implanta en el útero, no acaba de desarrollarse el embrión.
Se trata de un problema relativamente frecuente. Cerca de un 15 % de los embarazos que son detectados clínicamente acaban perdiéndose de forma espontánea, y en un tercio de ellos se debe a embarazos anembrionarios.
De este modo, la principal consecuencia para la mujer del embarazo anembrionario es que se producirá el aborto por la inviabilidad de la gestación.
Causas del embarazo anembrionario
La causa más habitual de los embarazos anembrionarios son las anomalías cromosómicas o genéticas que tienen lugar en el momento de la fecundación.
Algunas de las alteraciones cromosómicas más frecuentes son las trisomías, monosomías o poliploidías.
No existen factores relacionados con las características de la mujer o el hombre que puedan predecir con certeza que se producirá este tipo de gestación fallida. Sin embargo, los abortos espontáneos suelen ser más frecuentes en función de la edad de la madre. A partir de los 35 años se incrementa el riesgo de problemas en el embarazo. De igual modo, si se han producido abortos con anterioridad, el riesgo de nuevos abortos también es mayor.
Otros factores que muestran una posible asociación con los abortos espontáneos y los trastornos del embarazo están relacionados con niveles insuficientes de vitaminas del grupo B como el ácido fólico y vitamina K en la madre.
Síntomas del embarazo anembrionario y cómo detectarlo
En muchos casos el embarazo anembrionario puede darse sin síntomas y pasar totalmente desapercibido. Incluso, el aborto espontáneo puede expulsarse junto con la regla.
En el caso de realizar una prueba de embarazo, esta daría positivo, ya que la hormona detectada en dicha prueba es segregada por células del saco gestacional, aunque no aparezca un embrión en su interior.
En cuanto a los síntomas para la mujer en el transcurso de un embarazo anembrionario, estos serían los síntomas propios de un embarazo normal, incluyendo:
Ausencia de la menstruación.
Aumento de la sensibilidad mamaria.
Náuseas.
Etc.
Estos síntomas cesarían en el momento en el que tuviera lugar el aborto.
Por otro lado, la realización de una ecografía en etapas tempranas de la gestación podría ayudar a detectar un embarazo anembrionario.
La ecografía transvaginal permite detectar el embrión en torno a las 6 semanas de gestación. El diagnóstico de embarazo anembrionario se podrá establecer ante el hallazgo de un saco gestacional mayor de dos centímetros y en cuyo interior no se visualice el embrión.
¿El embarazo anembrionario puede ser recurrente?
No suele ser normal que se repita un embarazo anembrionario, aunque es posible que se pueda dar en más de una ocasión. Haber sufrido una gestación anembrionaria no supone un mayor riesgo de sufrir otra.
De este modo, se podría intentar una nueva gestación a los 2 o 3 meses desde que se produjera un embarazo anembrionario, en función de cómo se haya producido el proceso abortivo y de las indicaciones del ginecólogo.
En el caso de presentar varias pérdidas gestacionales, sería recomendable realizar un estudio para identificar los motivos que pudieran estar detrás de los abortos recurrentes e intentar ponerles remedio.

Artículo especializado
Desgarro perineal: ¿Cuándo se produce?
El parto vaginal se asocia con la posibilidad de desgarros perineales, afectando al 80-85% de las mujeres durante el parto.
¿Qué es el desgarro perineal?
El desgarro perineal es una solución de continuidad, esto es, un corte transversal que afectará a distintos músculos según su grado (asociado a la profundidad del corte).
De este modo, debido a la maniobra en la que la cabeza del bebé debe atravesar la abertura vaginal durante el parto, se puede producir una lesión en la zona perineal, ya sea un desgarro o una episiotomía realizada expresamente por el médico.
Además, el parto vaginal también se relaciona con lesiones de la musculatura perineal, del esfínter anal y lesiones del músculo elevador del ano, con mayor incidencia en el parto instrumental (parto a través de fórceps o ventosa) que en el parto espontáneo.
Grados de desgarro perineal
Los desgarros o traumas perineales se pueden clasificar en función de la profundidad de la región anatómica a la que afecten en:
Desgarros de primer grado. Afectan a la piel y mucosa vaginal.
Desgarros de segundo grado. Afectan a la musculatura, excluyendo el esfínter anal.
Desgarros de tercer grado (clasificación de Sultan):
a) Menos del 50% del espesor del esfínter anal externo.
b) Lesión del 50% o más del espesor del esfínter anal externo.
c) Lesión que afecta al esfínter anal externo y al interno.
Desgarros de cuarto grado. Desgarro del esfínter anal y la mucosa rectal.
Consecuencias del desgarro perineal
Cuando se produce un desgarro perineal es importante una correcta identificación del mismo, ya que de lo contrario pueden quedar secuelas como dispareunia o dolor durante el acto sexual, dolor perineal crónico, incontinencia urinaria o incontinencia fecal.
En el caso de la lesión del esfínter anal durante el parto, su relevancia radica en su relación con la incontinencia anal y con diferentes síntomas defecatorios, incluyendo la urgencia defecatoria y el dolor.
La incontinencia anal se define como la pérdida involuntaria de gases o heces, que suele estar precedida de una sensación de urgencia y que puede afectar de forma muy importante a la calidad de vida de la mujer.
Prevención, tratamiento y alivio del desgarro perineal
Es importante tener en cuenta durante el parto las actuaciones necesarias para prevenir el desgarro perineal durante el parto. De este modo, se pueden establecer las siguientes recomendaciones:
Proteger el periné . Para ello, se debe aprovechar la elasticidad del suelo pélvico y lograr la expulsión de la cabeza del bebé en máxima flexión.
Aplicación de compresas calientes y masaje perineal. Con la intención de aumentar la flexibilidad y reducir la tensión de la musculatura perineal (se recomienda a partir de la 33 semana de embarazo).
Uso restrictivo de la episiotomía . Se aconseja usar la episiotomía mediolateral derecha en lugar de la central. Esta debe tener un ángulo de al menos 60° para que sea protectora (por cada 6° que se horizontalice la incisión de la episiotomía, disminuye un 50% el riesgo de desgarros con peores consecuencias).
Entre los factores de riesgo para la aparición de lesiones perineales más graves (tercer y cuarto grado) se encuentran:
No haber tenido hijos anteriormente.
Peso fetal superior a 4 kg.
Parto instrumentado con fórceps.
Distocia de hombros.
Episiotomía media.
Presentación occípito-posterior.
Etapa expulsiva del parto superior a una hora.
Posición materna en litotomía o en cuclillas.
Entre estos factores, algunos son modificables, sobre todo la instrumentación del parto, la realización de episiotomía y la posición materna. Su modificación permite disminuir la incidencia de las lesiones anales.
Por otro lado, la episiotomía sistemática no ha demostrado ser útil para la prevención de los desgarros perineales, por esto se aconseja realizarla de forma restrictiva.
En cuanto a la reparación de los desgarros perineales, se recomienda su sutura una vez que se ha producido el parto, salvo que se trate de lesiones del periné anterior que no sangren ni distorsionen la anatomía y de la piel del periné o el epitelio vaginal si los bordes están próximos y no hay sangrado.
Finalmente, en algunos casos es recomendable algún tratamiento farmacológico. Es el caso de una profilaxis antibiótica intravenosa antes de la reparación del desgarro del esfínter anal y su prolongación durante 5-7 días.
También se puede establecer un tratamiento con laxantes en caso de desgarro del esfínter anal o de la mucosa rectal.
Desgarro perineal y embarazo
En el caso de mujeres que han tenido un desgarro perineal, especialmente de los grados más graves, a la hora de plantearse otro embarazo la preocupación se centra en la posibilidad de que esta lesión pueda repetirse y que aparezcan o se agraven los síntomas.
De este modo, después de haber padecido un desgarro de tercer o cuarto grado durante el parto, si la mujer se vuelve a quedar embarazada, se debería tener en cuenta:
Si la mujer está asintomática y mantiene la continencia, se puede recomendar un parto vaginal. El riesgo de repetir otra lesión de tercer o cuarto grado es relativamente baja. En cualquier caso, la incontinencia anal no se modifica por la realización de una cesárea. En el caso de pesos fetales por encima de 4 kg el riesgo de lesión aumenta notablemente.
Si la mujer presenta incontinencia anal, es probable que el embarazo aumente la intensidad de la sintomatología. Sin embargo, no hay evidencia de que la vía de parto influya en la evolución posterior.
En los casos en que la mujer haya presentado una incontinencia anal posparto, con cirugía exitosa posterior, es recomendable el parto por cesárea .

Artículo especializado
El hierro en el embarazo y su importancia
La dieta de la embarazada debe contener un aporte energético adecuado para asegurar su propia salud y la del feto, ya que, desde el punto de vista nutricional, la dependencia del feto del organismo materno es total.
El hierro es uno de los nutrientes importantes durante el embarazo debido al elevado riesgo de anemia en esta etapa, dada la mayor demanda del mismo por parte de la gestante. En general, los niveles de hierro del feto dependen de los niveles de la madre, siendo un elemento esencial para el desarrollo de la mayoría de los órganos en el feto y también para el desarrollo normal del cerebro.
Anemia en el embarazo
La anemia ferropénica es muy frecuente en las mujeres embarazadas, sobre todo en el segundo y tercer trimestre de la gestación y después del parto. Algunas estimaciones señalan que el déficit de hierro durante el embarazo afecta a una de cada cinco embarazadas, pudiendo llegar a una de cada tres durante el tercer trimestre de embarazo.
Por otro lado, en España el déficit de hierro supone el 90% del total de anemias en las gestantes. De este modo, muchas gestantes necesitarán suplementarse con hierro durante esta etapa, si bien una alimentación adecuada puede prevenir la anemia por deficiencia de hierro en el transcurso de la gestación.
La Sociedad Española de Ginecología y Obstetricia (SEGO) recomienda la suplementación de hierro desde el segundo trimestre hasta pasado un mes del parto.
Se debe asegurar el aporte de 30 mg de hierro al día durante el embarazo en las gestaciones únicas y 60 mg/día en las gestaciones múltiples. Durante la lactancia el aporte debe ser de 15 mg/día.
Alimentos con hierro para el embarazo
Una dieta adecuada durante el embarazo debe incluir alimentos ricos en hierro, como carnes rojas magras, carne de ave, pescado y moluscos. Además, las opciones vegetales comprenden sobre todo las legumbres y verduras como espinacas y acelgas y los frutos secos.
El hierro que proviene de los productos de origen animal (hierro hemo), se absorbe mejor que el de origen vegetal. Para aumentar la absorción de hierro de las fuentes vegetales y de los suplementos es recomendable consumirlos junto con alimentos ricos en vitamina C (naranjas, tomates, fresas, kiwi, pimientos, etc.).
Para poder prevenir y tratar la anemia ferropénica, Savia pone a tu disposición los mejores especialistas en Endocrinología y Nutrición .
Síntomas de hierro bajo en el embarazo
Los síntomas de unos niveles bajos de hierro sin llegar a anemia son inespecíficos. La disminución de las reservas corporales de hierro puede dar lugar a la aparición de fatiga.
Por otro lado, cuando se desarrolla anemia por deficiencia de hierro (anemia ferropénica), los síntomas son más específicos y pueden aumentar con la severidad de la anemia.
En cualquier caso, los síntomas de la anemia, sobre todo en casos leves, pueden ser muy parecidos a los síntomas generales que se experimentan por el propio embarazo. De este modo, una mujer embarazada con niveles bajos de hierro puede experimentar:
Cansancio y debilidad.
Palidez de la piel y las mucosas.
Mareos.
Deterioro de la capacidad cognitiva.
Inestabilidad emocional y síntomas depresivos.
Mayor riesgo de infecciones.
En casos más graves, pueden aparecer palpitaciones, dificultad para respirar y dolor en el pecho.
Los análisis de hierro y hemoglobina, la principal proteína que lo contiene, serán habituales a lo largo del embarazo, junto con otros controles y análisis .
Síntomas de hierro alto en el embarazo
Tomar demasiado hierro durante el embarazo también puede ser contraproducente para la madre y el feto.
Algunos de los efectos relacionados con un exceso de hierro incluyen mayor riesgo de:
Preeclampsia.
Hipertensión arterial.
Diabetes mellitus.
Abortos espontáneos.
Consecuencias de la anemia en el embarazo
La anemia por deficiencia de hierro durante la gestación puede tener consecuencias negativas para la salud de las mujeres, impidiendo la realización normal de sus actividades cotidianas.
Las formas más graves de anemia pueden tener una influencia negativa en la gestación, ya que existe un mayor riesgo de aborto y de parto pretérmino, bajo peso al nacer (provocando, a su vez, un aumento de la mortalidad perinatal), así como un incremento del riesgo de infecciones puerperales.
Adicionalmente, la deficiencia de hierro en las madres, especialmente a principios del embarazo, está significativamente asociada con parto prematuro, peso bajo del recién nacido, peso bajo para la edad gestacional y una mayor mortalidad perinatal del neonato.
La deficiencia de hierro y la anemia durante la gestación inevitablemente se agravarán después de dar a luz (anemia posparto), debido a las pérdidas de sangre asociadas con el parto. Se estima que la prevalencia de anemia en el posparto es del 50% dentro de las 48 horas siguientes al parto. La mayoría de las formas serían prevenibles con una gestante que llegase al último trimestre de la gestación con sus reservas férricas en óptimas condiciones (hemoglobina mayor o igual a 11mg/dl).
Por otro lado, la deficiencia de hierro en la gestante afectará negativamente las interacciones entre la madre y el niño. Además, los niños nacidos de madres con deficiencia de hierro tienen un menor desarrollo cognitivo, motor, social, emocional y neurofisiológico de las funciones cerebrales.
En definitiva, después de analizar los resultados de los análisis correspondientes, el ginecólogo podrá recomendar la necesidad de suplementos de hierro y vitaminas, algunas de ellas implicadas en el metabolismo del hierro.

Artículo especializado
Cuerpo lúteo: ¿Qué es y cuándo se forma?
El ciclo menstrual dura de media 28 días, si bien existen diferencias entre mujeres y la duración puede variar con la edad.
Este ciclo se divide en varias fases, que incluyen el sangrado o menstruación, seguida de la fase folicular en la que se desarrolla el futuro óvulo, la ovulación y finalmente el cuerpo lúteo , que antecede a un nuevo sangrado o menstruación.
¿Qué es el cuerpo lúteo?
El cuerpo lúteo es el tejido formado después de que el óvulo sea liberado desde el folículo ovárico. Una de sus principales funciones es la producción de progesterona para el potencial establecimiento y mantenimiento de la gestación.
Cuando se libera la hormona folículo estimulante (FSH) en el cerebro, en el ovario se comienzan a desarrollar folículos, en cuyo interior se forman los óvulos. El folículo dominante conseguirá alcanzar un mayor desarrollo frente al resto de folículos, liberando el óvulo en ese ciclo menstrual. Además, los folículos se encargan de liberar estradiol que provocará en el útero el engrosamiento del endometrio.
Una vez que la ovulación ha tenido lugar, el folículo que lo contenía se transforma en cuerpo lúteo (también conocido como cuerpo amarillo).
La doble función del cuerpo lúteo incluye el mantenimiento de la actividad menstrual en los ciclos en los que no hay fecundación y, cuando esta sí tiene lugar, promueve la liberación de sustancias endocrinas que permiten la iniciación y el avance de la gestación.
¿Cuándo se forma el cuerpo lúteo?
Tras la ovulación se inicia la fase lútea, en la que se desarrolla el cuerpo lúteo. Esta fase se extiende aproximadamente desde el día 15 al 28 del ciclo menstrual.
El cuerpo lúteo perdurará hasta que se produzca una posible fecundación del óvulo o tenga lugar la menstruación, con el comienzo de un nuevo ciclo menstrual.
Tras la ovulación, el folículo va disminuyendo de tamaño mientras sigue produciendo progesterona y estrógenos que preparan al útero para recibir a un posible embrión.
Tras la liberación del óvulo por el cuerpo lúteo, este se dirige hasta el útero a través de la trompa de Falopio. Cuando se produce la fecundación del óvulo, el embrión se implanta una semana después de la fecundación. En ese momento el organismo comienza a producir la gonadotropina coriónica humana (hCG) encargada de mantener el folículo activo en su producción de estrógenos y progesterona. Con ello se evitará que se desprenda el revestimiento del útero, formándose y madurando la placenta donde tendrá lugar la mayor parte del embarazo.
Por el contrario, si no tiene lugar la fecundación, el folículo se va contrayendo lo que determina una disminución de los niveles de estrógenos y progesterona. Esto dará lugar a que el revestimiento del útero se desprenda y se produzca la hemorragia de la menstruación.
¿Qué hormona se produce en el cuerpo lúteo?
Como se ha dicho, el cuerpo lúteo secreta progesterona y estrógenos. Cuando se produce la fecundación del óvulo el cuerpo amarillo se hace más grande y su producción de hormonas se incrementa para sustentar el embarazo. Cuando la fecundación no tiene lugar, encoge y degenera, disminuyendo su producción hormonal.
¿Cuánto tiempo dura el cuerpo lúteo?
La fase lútea tiene una duración de 14 días que puede fluctuar en 2 días más o menos. Después de la ovulación, la estructura folicular que queda en el ovario convirtiéndose en el cuerpo lúteo, asume su función de glándula liberadora de hormonas. La vida del cuerpo lúteo hace que su degeneración a los 14 días determine el fin del ciclo menstrual. De este modo, al final de la fase lútea los niveles de progesterona y estrógeno disminuyen, dando lugar a la menstruación.
Por otro lado, aunque el día más común de la ovulación es el día 15 del ciclo menstrual (que en muchos casos no dura 28 días), el momento de la ovulación también puede presentar una gran variabilidad en el tiempo. Además, la duración de la fase lútea puede variar dentro de una misma mujer de un ciclo a otro. De este modo, una mujer puede ovular en un día del ciclo diferente en cada ciclo y, de igual modo, entrar en la fase lútea también en días diferentes.
Artículo especializado
PH vaginal: cómo regularlo
Las infecciones vulvovaginales son un motivo muy frecuente de consulta médica, estimándose que el 75% de las mujeres experimentan un episodio de vulvovaginitis a lo largo de su vida. Uno de los principales factores relacionados es el pH vaginal.
¿Qué es el pH vaginal?
El pH es un parámetro que indica el grado de acidez-alcalinidad de un medio y su valor puede oscilar entre 0 (máxima acidez) y 14 (máxima alcalinidad). En la vagina el pH óptimo (o la acidez vaginal) se sitúa entre 3,5 y 4,5. Sin embargo, se trata de un valor que puede experimentar variaciones en función de diferentes factores, incluyendo la edad de la mujer, el embarazo, la menopausia o cuando se producen infecciones.
¿Cuál es la función del pH vaginal?
El pH vaginal adecuado ayuda a mantener sana la mucosa vaginal y proteger frente a posibles infecciones.
En condiciones normales, existe una colonia de microorganismos en la vagina que constituye su microbiota o flora natural. Cuando el pH vaginal se desequilibra se puede dar lugar a la proliferación de microorganismos patógenos que causen infecciones y dolencias. En la flora vaginal normal abundan los lactobacilos, un tipo de bacterias que se caracterizan por su efecto protector frente a diferentes agentes patógenos.
De este modo, cuando tiene lugar un pH vaginal alterado la población de lactobacilos se puede ver desplazada junto con su efecto protector. Esta situación puede dar lugar a la aparición de patologías infecciosas a nivel vaginal como las candidiasis, la vaginosis bacteriana y la tricomoniasis.
Microbiota vaginal normal: la importancia de los lactobacilos
La alteración de la microbiota vaginal es la causa principal de las infecciones vaginales.
La microbiota vaginal es el conjunto de microorganismos que habitan de manera natural y sin causar daño en la región vaginal. Su composición cambia notablemente con la edad, dependiendo de las variaciones en los niveles de estrógenos.
De este modo, cuando aparecen los ciclos menstruales, el epitelio vaginal aumenta su grosor, segregando un exudado que contiene glucógeno y otros nutrientes. Este hecho facilita la colonización principalmente de lactobacilos, pero también de otras bacterias como Gardnerella vaginalis y hongos como Candida albicans . En un momento determinado estos microorganismos pueden convertirse en patógenos si proliferan en exceso o se produce una modificación en el pH vaginal.
Los lactobacilos son los principales garantes del mantenimiento del ecosistema vaginal, promoviendo mecanismos como:
Competición con los hongos por los nutrientes disponibles.
Bloqueo de receptores epiteliales para los hongos.
Potenciación de la respuesta inmune.
Generación de sustancias capaces de producir ácido láctico a partir de la glucosa (peróxido de hidrógeno, lactacinas y acidolinas). El ácido láctico es el responsable de mantener el pH vaginal en rangos entre 3,5 y 4,5. Este constituye el principal mecanismo de defensa frente a la colonización por patógenos.
Los lactobacillus metabolizan el glucógeno secretado por las células más superficiales de la mucosa vaginal y, de esta forma, contribuyen a mantener unos niveles de pH adecuados. Cuando tiene lugar la menopausia, se produce una disminución, incluso desaparición, de la capa de células superficiales, dando lugar a una disminución en la presencia de Lactobacillus, alteraciones en el pH y a la proliferación de microorganismos patógenos.
¿Qué valores de pH vaginal son normales?
A lo largo de la vida de una mujer el pH vaginal va experimentando una evolución que determina diferentes valores de normalidad. De este modo, durante la infancia y hasta la pubertad el pH de la vagina es neutro (cercano a 7). Posteriormente, el aumento de estrógenos y la instauración de la microbiota normal durante la edad reproductiva determinan una disminución de los valores de pH (entre 3,5 y 4,5). Finalmente, tras la menopausia el pH vaginal vuelve a la neutralidad.
Regular el pH vaginal puede contribuir a evitar infecciones y la aparición de síntomas molestos como irritación, picor, enrojecimiento y sequedad.
Síntomas del desequilibrio del pH vaginal
Como se ha dicho, el desequilibrio del pH vaginal puede dar lugar a la modificación de la flora vaginal y a la posible proliferación de microorganismos patógenos. Esto puede ir asociado a síntomas de mayor o menor intensidad entre los que se encuentran:
Modificaciones del flujo vaginal (cambios de color, olor y consistencia).
Picor y escozor.
Sequedad vaginal.
Molestias al orinar.
Molestias durante las relaciones sexuales.
Ante la aparición de estos síntomas es recomendable consultar con un especialista en Ginecología y Obstetricia.
Factores que pueden alterar el pH vaginal
El equilibrio del pH vaginal depende de una serie de factores, algunos de los cuales pueden ser controlados de forma individual modificando ciertos hábitos. Entre estos factores se encuentran:
Higiene vaginal . En ocasiones la utilización de jabones agresivos puede irritar el interior de la vagina, modificando el pH. Esto contribuye a destruir la flora vaginal normal y propiciando la proliferación de microorganismos patógenos.
Productos perfumados . Ciertas sustancias aromáticas presentes en compresas, papel higiénico, etc., no son adecuados para mantener un pH vaginal normal.
Tratamiento con antibióticos . Los antibióticos utilizados de forma prolongada o frecuente pueden afectar negativamente a la flora vaginal.
Por otro lado, la gestación, la lactancia o la menopausia son etapas en la vida de la mujer en las que, debido a las variaciones hormonales características de cada una de ellas, pueden tener lugar alteraciones en el pH vaginal.
Consecuencias del desequilibrio del pH vaginal
La vulvovaginitis es la inflamación de la mucosa vaginal y de la piel vulvar. La vulvovaginitis causada por el hongo Candida es la más prevalente, aunque también pueden producirse infecciones por bacterias.
La vulvovaginitis candidiásica es una enfermedad inflamatoria de la vagina, producida por diferentes especies de hongos, fundamentalmente de Candida −siendo la más frecuente dentro de esta especie la Candida Albicans − que tiene lugar en condiciones fisiológicas alteradas con disminución de la inmunidad local. Se trata de un proceso muy común en las mujeres adultas, con un pico máximo de incidencia entre los 20 y 40 años. Se calcula que, a los 25 años el 50% de las mujeres habrá tenido al menos un episodio. De igual modo, entre las mujeres premenopáusicas el 75% habrá sufrido al menos un episodio y el 45% dos episodios o más de vulvovaginitis candidiásica.
Las alteraciones en la inmunidad leves que se producen previas a tener la menstruación también pueden favorecer la aparición de candidiasis vaginal.
Por otro lado, las vaginosis bacterianas son menos frecuentes y se caracterizan por un incremento de la secreción vaginal, que se hace más acuosa y maloliente, y que se acompaña de escasa sintomatología adicional. El mal olor de este flujo se suele poner más de manifiesto cuando entra en contacto con fluidos de pH alcalino (como el semen, la sangre menstrual y el gel empleado en exploraciones ginecológicas, entre otros).
También se puede producir una inflamación vulvar o vaginal de origen irritativo o alérgico que no esté causada por ningún microorganismo. Estas suelen estar relacionadas con la utilización de jabones o cremas irritantes, el empleo de ropa interior de tejidos sintéticos y al uso frecuente de protectores (compresas, salvaslips, pañales, etc.).
Artículo especializado
Hormona hcg: ¿Qué es y cuál es su función en el embarazo?
La hormona gonadotropina coriónica humana (u hormona hCG por sus siglas en inglés) es una hormona que se produce exclusivamente durante el embarazo.
¿Qué es la hormona hcg?
La hormona hCG es una glucoproteína que es liberada por el embrión cuando este se implanta en el útero materno. De este modo, la hCG también es conocida como “hormona del embarazo” y sirve para confirmar que la fecundación ha tenido lugar y se da inicio al embarazo.
La hormona hCG está formada por dos subunidades o fracciones:
Alfa-hCG . Se trata de una parte común a otras hormonas como la TSH (hormona estimulante de la tiroides), la FSH (hormona folículo estimulante) o la LH (hormona luteinizante).
Beta-hCG . Es exclusiva de la hormona hCG y es la que se utiliza en los test de embarazo.
A la hormona hCG se le atribuyen algunos síntomas característicos del embarazo , como las náuseas y los vómitos. Durante el primer trimestre de la gestación tiene lugar el mayor aumento de la hormona, por lo que es en ese periodo cuando suelen aparecer más los síntomas. La hCG también se relaciona con otros síntomas como el cansancio y la sensación de sueño, la irritabilidad, los problemas gastrointestinales y los cambios de humor.
Utilidad clínica de la determinación de hCG
La determinación de la hCG en la orina y la sangre es de gran utilidad clínica en el diagnóstico del embarazo normal, pero también de sus patologías, como el embarazo ectópico, embarazo anembrionado y la muerte del embrión.
También puede ser útil en la detección de trisomías, determinadas patologías gestacionales, tumores ováricos, en la inducción médica de la ovulación y el control de la fertilidad, entre otras situaciones.
¿Cuántos mUI/ml debo tener para estar embarazada?
La hormona hCG está presente en el organismo de la mujer durante toda la gestación, si bien sus valores varían conforme evoluciona el embarazo.
Durante el primer trimestre del embarazo la hCG aumenta hasta llegar a un pico de concentración máxima en la semana 12-14 de gestación.
La hormona coriónica humana en el embarazo normal incrementa sus valores en las primeras semanas de una manera casi constante, duplicando su valor cada 48 horas. Sus concentraciones en orina y sangre se correlacionan muy bien con la cantidad de tejido que envuelve y protege al embrión (el trofoblasto). De este modo, los niveles de hCG permiten el seguimiento del embarazo en sus primeras fases.
En los embarazos ectópicos o extrauterinos, en los que existe poco tejido trofoblástico, los niveles en sangre y orina de la hormona serán más bajos.
Por el contrario, las elevadas concentraciones de hCG por encima de los valores normales pueden hacer sospechar de la presencia de un embarazo múltiple.
La combinación de ultrasonografía transvaginal, junto con la determinación cuantitativa de hCG puede ayudar a establecer, de forma temprana, diagnósticos sobre las gestaciones ectópicas.
Los intervalos orientativos de referencia para la hormona hCG en sangre en función de las semanas de embarazo se pueden cifrar en:
3 semanas de embarazo: de 5 a 50 mUI/ml.
4 semanas: entre 5 y 130 mUI/ml.
5 semanas: entre 75 y 7.000 mUI/ml.
6 semanas: entre 1.080 y 56.500 mUI/ml.
Entre 7 y 8 semanas: entre 7.650 y 229.000 mUI/ml.
Entre 9 y 12 semanas: entre 25.700 y 288.000 mUI/ml.
Entre 13 y 16 semanas: entre 13.300 y 254.000 mUI/ml.
Entre 17 y 24 semanas: entre 4.060 y 165.400 mUI/ml.
Entre 25 y 40 semanas: entre 3.640 y 117.000 mUI/ml.
En cualquier caso, las concentraciones de hCG en suero y orina varían sustancialmente durante el embarazo y entre diferentes mujeres.
¿Cómo se detecta la hormona gonadotropina coriónica?
Las pruebas de embarazo (o prueba hCG) están basadas en la detección de la fracción beta de la hCG. Estas pueden ser realizadas en orina o en sangre. Pueden distinguirse dos tipos de pruebas:
Test de embarazo cuantitativos . Señalan los niveles exactos de la hormona en la sangre materna. Estas pruebas permiten establecer unos valores de referencia según las semanas de gestación.
Test de embarazo cualitativos . Indican la presencia o ausencia de la hormona en sangre o en orina y, por tanto, la existencia o no de embarazo.
Cuando se desea saber la existencia o no del embarazo, lo ideal es esperar para realizar el test de embarazo hasta el primer retraso de la menstruación o 15 días después de haberse producido la relación sexual.
Para hacer la prueba de embarazo con resultados fiables en el transcurso de un tratamiento de reproducción asistida, se recomienda esperar unos 10-15 días desde la transferencia embrionaria o la inseminación artificial.
¿Cómo produce la hormona gonadotropina coriónica?
La hormona gonadotropina coriónica humana (hCG) es una proteína sintetizada principalmente por los tejidos embrionarios.
Su secreción se relaciona con la cantidad de tejido trofoblástico que envuelve el embrión desde la semana 4 a la 20 de embarazo. A partir de la semana 20 los niveles se relacionan con el peso del feto.

Artículo especializado
Absceso mamario: ¿Qué es y cómo tratarlo?
Los efectos beneficiosos de la lactancia materna en la salud de la madre y el bebé son ampliamente conocidos, recomendándose de manera exclusiva durante los seis primeros meses de vida y su continuación durante los siguientes meses de vida del bebé. Sin embargo, muchas mujeres dejan de amamantar debido a la mastitis y los problemas derivados de la aparición de abscesos mamarios.
¿Qué es el absceso mamario?
Cuando se produce una mastitis o inflamación del tejido mamario, esta puede estar asociada a un proceso infeccioso que suele coincidir con el periodo de lactancia.
En estas circunstancias también puede tener lugar la aparición de un absceso en el pecho o absceso mamario , consistente en una acumulación de líquido infectado o pus en el tejido mamario. En la mayoría de las ocasiones, este se origina por una mastitis que no ha sido tratada a tiempo o no de manera adecuada. El absceso mamario suele ser una acumulación localizada. Las principales bacterias causantes de los abscesos suelen ser de tipo anaerobio que necesitan un medio sin oxígeno para sobrevivir.
Por otro lado, un absceso de mama sin lactancia es aquel que tienen lugar cuando la mujer que lo padece no está amamantando o en periodo de embarazo.
Habitualmente los abscesos se tratan con antibióticos, incisión y drenaje o aspiración del líquido por medio de una aguja guiada por ecografía.
Los abscesos mamarios son un importante problema para la salud de la mujer, con tendencia a ser recurrentes. Un factor de riesgo importante para la recurrencia es que la mujer sea fumadora. Además, pueden dejar secuelas permanentes, como deformidades en la mama o la disminución de la capacidad de producir leche.
¿Por qué se produce el absceso mamario?
La mayor parte de los abscesos mamarios suelen ser consecuencia de una complicación cuando se produce una mastitis o inflamación de la mama. Esta tiene lugar generalmente por una infección bacteriana que afecta al seno.
La mastitis no es exclusiva de las mujeres lactantes, pero suele relacionarse con el periodo en el que la madre amamanta a su hijo.
Infecciones bacterianas
La mayoría de los abscesos mamarios tienen su causa en una infección de origen bacteriano.
Las bacterias causantes de la infección pueden acceder al interior de la mama a través de pequeñas roturas o grietas que se producen en el pezón producidas debido a la fricción de la lactancia.
También pueden proliferar en los conductos mamarios cuando la leche se acumula y se queda estancada por la obstaculización de un conducto. Ocasionalmente, la infección puede producirse por vía hematógena.
La proliferación bacteriana que se produce es respondida por el sistema inmunitario, creándose un pequeño acúmulo de bacterias y leucocitos muertos (pus). De este modo, el objetivo es contener la infección y reducirla a una localización, intentando evitar de esta forma la diseminación a otros lugares del organismo.
El microorganismo más frecuentemente implicado en el absceso mamario es el Staphylococcus aureus , seguido por Staphylococcus epidermidis .
Factores de riesgo
La aparición de abscesos mamarios suele darse con mayor frecuencia en determinadas circunstancias:
En mujeres durante su primer embarazo.
Edad de la madre superior a los 30 años.
Gestaciones largas.
Obesidad o diabetes de la madre.
Mujeres que trabajan fuera del hogar.
Mujeres fumadoras.
Si existen antecedentes de mastitis.
Cómo detectar el absceso mamario
El absceso mamario resulta muy perjudicial durante la lactancia, aunque a veces es difícil de detectar. La incidencia del absceso mamario como complicación de una mastitis se estima entre el 3 y el 11%.
Este debe sospecharse en caso de progresión de una mastitis o infección mamaria y persistencia de una zona bien definida con los siguientes síntomas, a pesar de que ya pueda existir un tratamiento en curso:
Enrojecimiento
Dolor fluctuante.
Fiebre
Ganglios linfáticos aumentados.
El signo más característico del absceso es la aparición de una induración que resulta fluctuante al tacto. Pero no siempre está presente, sobre todo en abscesos localizados en regiones profundas no accesibles a la exploración física mediante palpación.
De este modo, la detección de los abscesos mamarios también puede estar condicionada a su localización. Esta puede ser:
Superficial . La zona afectada coincidente con la de máximo enrojecimiento.
Profunda . La localización interna determina la aparición tardía de los síntomas inflamatorios, con un mayor retraso en el diagnóstico, lo que puede provocar un mayor tamaño del absceso y una mayor destrucción de tejidos mamarios.
Diagnóstico del absceso mamario
El diagnóstico está basado en los síntomas y signos. Si es necesario se pueden utilizar pruebas complementarias como la ecografía y la punción con aspiración por medio de aguja fina.
La ecografía puede servir para diferenciar la mastitis difusa y el absceso mamario localizado.
Finalmente, el cultivo de la leche materna es esencial para el correcto diagnóstico microbiológico y el establecimiento del tratamiento más adecuado.
¿Qué hacer si tenemos un absceso mamario?
Ante la presencia de síntomas de mastitis o absceso mamario es fundamental acudir al ginecólogo.
Si se confirma la presencia de un absceso, el tratamiento se basará en el drenaje y la administración de antibióticos. La selección de los medicamentos antibióticos específicos para los abscesos mamarios debe estar dirigida a las bacterias que con más frecuencia producen estas infecciones y en función de los resultados del cultivo de la leche materna. Ayudará a establecer un tratamiento antibiótico efectivo la realización de un antibiograma para establecer las resistencias específicas de los microorganismos desarrolladas frente a los diferentes fármacos.
Medidas preventivas
Para la prevención de la aparición de abscesos es preciso asegurar un adecuado vaciamiento del pecho para evitar el estancamiento de la leche en los conductos mamarios y tratar de forma precoz el llenado excesivo de las mamas (ingurgitación mamaria), así como las grietas en los pezones y el bloqueo de los conductos mamarios.
Algunos consejos que pueden ser útiles incluyen:
Ofrecer el pecho a demanda sin límites en la duración de la toma.
Extracción de la leche restante tras la toma si el vaciado no ha sido completo.
Evitar la compresión de la mama (por ejemplo, con el uso de ropa ajustada).
Procurar un descanso materno adecuado.
Medidas de higiene de manos de la mujer y las personas que puedan contactar con la mama.
Tratamiento precoz de las grietas infectadas en la mama con antibióticos de uso tópico. La mejor prevención para la aparición de grietas en la mama es una correcta técnica de lactancia. Por tanto, es muy importante el asesoramiento y el acompañamiento por personal especializado en esta etapa.
También es importante una adecuada educación materna en el ámbito de la lactancia, incluyendo aspectos como:
Correcta extracción de la leche.
Exploración de la mama con identificación de ingurgitación, estasis e inflamación mamaria.
Ante cualquier duda sobre la presencia de infección o absceso mamario es preciso consultar con el especialista en Ginecología y Obstetricia.
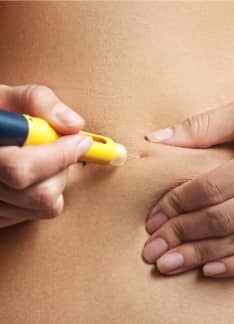
Artículo especializado
Estimulación ovárica: ¿En qué consiste?
La estimulación ovárica es un tratamiento médico que tiene el objetivo de estimular los ovarios, alcanzando así el desarrollo de varios folículos maduros que permitan conseguir el número máximo de ovocitos. En general, la estimulación ovárica es el primer tratamiento al que se somete la mujer −en el contexto de la reproducción asistida− permitiendo resolver los trastornos de ovulación que pudieran existir e incrementando la calidad y la cantidad de los óvulos que se pueden generan.
¿Qué es la estimulación ovárica?
La estimulación ovárica consiste en la administración de hormonas femeninas inyectables capaces de inducir el crecimiento folicular múltiple con el objetivo de obtener un mayor número de óvulos viables. Este procedimiento se inicia entre el primer y tercer día de la menstruación.
Las hormonas administradas son gonadotropinas exógenas: la hormona folículo estimulante (FSH) y la hormona luteinizante (LH). Ambas por vía subcutánea y en dosis superiores a las que se producen de manera natural en el organismo. De este modo, el aumento de la concentración plasmática de FSH y LH, afectarán tanto a la cantidad como a la calidad de los ovocitos obtenidos.
En este sentido, el número de ovocitos obtenidos está relacionado con la dosis inicial de gonadotropinas administrada. Aun así, se pueden producir variaciones en función de las características individuales de cada paciente, como pueden ser la edad, los resultados de posibles ciclos previos de estimulación para reproducción asistida y de los datos de la estimación de la reserva ovárica.
La dosis inicial más habitual en pacientes con una expectativa de respuesta normal es de 150-225 unidades internacionales. En cualquier caso, para optimizar la estimulación ovárica se debe encontrar un equilibrio entre el resultado que se desea conseguir con el mínimo riesgo para la paciente, evitando la sobreestimulación ovárica.
¿En qué procesos se aplica la estimulación ovárica?
Cuando tener un hijo no es un proceso fácil y se dan casos de infertilidad y trastornos de la ovulación, muchas parejas se ven obligadas a recurrir a las técnicas de reproducción asistida para lograr el embarazo. Con el fin de incrementar las probabilidades de éxito es necesario someterse a la estimulación ovárica, para aumentar el número de ovocitos que se generan en la ovulación.
La estimulación ovárica suele incluirse en casi todos los tratamientos de fertilidad, siendo especialmente efectivo cuando la edad de la madre es elevada (a partir de los 35 años la función ovárica se ve disminuida), principal factor relacionado con una menor calidad y cantidad de óvulos.
Además, la estimulación ovárica se suele utilizar para obtener un mayor número de ovocitos en casos de donación o cuando la mujer opta por la congelación si desea retrasar la maternidad.
¿En qué consiste la estimulación ovárica natural?
La estimulación ovárica solo es posible mediante la utilización de hormonas de la ovulación y a través de inyecciones. No existe un método más natural. La administración, aunque es mediante inyecciones, es sencilla y no duele.
Inicialmente se utilizaban gonadotropinas menopáusicas urinarias que contenían concentraciones variables de hormona luteinizante y hormona folículo estimulante. Posteriormente se desarrollaron la FSH y LH recombinantes, lo que aportó fármacos con dosis constantes y de mayor disponibilidad.
Por otro lado, la fecundación in vitro natural es aquella en la que no se lleva a cabo una estimulación ovárica, por lo que el número de óvulos disponibles para su posterior extracción será menor.
Tratamiento de la estimulación ovárica
En la actualidad se dispone de un gran arsenal de fármacos inductores de estimulación ovárica, siendo las gonadotropinas (FSH y LH) los más empleados dentro de la técnica de reproducción in vitro.
La individualización del tratamiento de estimulación ovárica permite la predicción de la respuesta ovárica a la acción de las gonadotropinas. Este modelo tiene por objeto conseguir el mejor resultado posible en tasas de embarazo e hijo vivo, equilibrando los riesgos derivados de la intervención farmacológica como la hiperestimulación, así como los riesgos de cancelación del ciclo por baja respuesta.
La respuesta a la estimulación ovárica dependerá de la reserva ovárica, de la variabilidad funcional ovárica mensual y de las características de estimulación que se lleve a cabo. Normalmente se considera deseable una respuesta adecuada para obtener de 5 a 15 ovocitos (alcanzando las mayores tasas de embarazo en torno a los 15 ovocitos), ya que respuestas superiores o inferiores suelen provocar la finalización del tratamiento.
En cualquier caso, la decisión sobre el tipo de estimulación, las gonadotropinas y las dosis dependerá del ginecólogo, en función de las características de la pareja y siempre teniendo en cuenta su opinión.

Artículo especializado
La coenzima Q10 en el embarazo
La coenzima Q10 (CoQ10) es una sustancia presente de forma natural en las células humanas. Entre otras muchas acciones, se relaciona con beneficios para la fertilidad y para diferentes aspectos del embarazo.
Qué es la coenzima Q10
La coenzima Q10 −o ubiquinona, nombre que recibe por estar presente en multitud de tejidos− es un compuesto presente en la mayoría de las células, cuya principal característica es su naturaleza antioxidante que contribuye al crecimiento y mantenimiento celular.
La principal función de la coenzima Q10 en el organismo es promover el crecimiento celular y la protección de las células. Además, son necesarias cantidades adecuadas de CoQ10 para la respiración y obtención de energía a nivel celular. En las células que tienen altos requerimientos de energía es dónde se encuentras las mayores concentraciones de coenzima Q10, incluyendo las células del corazón, hígado y riñón.
En condiciones normales de salud, la CoQ10 no necesita ser suministrada a través de suplementos, ya que el organismo se encarga de sintetizar las cantidades suficientes de esta coenzima. No obstante, uno de los factores que determinan la disminución de los niveles de CoQ10 del organismo es el paso del tiempo. De este modo, el envejecimiento es una de las principales causas de presentar niveles disminuidos de coenzima Q10, haciendo necesario el consumo de suplementos.
Por otro lado, la CoQ10 se encuentra en diferentes alimentos (principalmente carnes, pescados y frutos secos), aunque en cantidades escasas como para aumentar significativamente los niveles de CoQ10 en el organismo.
En el curso de numerosos procesos patológicos, en los que existe una deficiencia de CoQ10, puede beneficiarse de su suplementación, incluyendo enfermedades mitocondriales, fibromialgia, enfermedades cardiovasculares y neurodegenerativas, cáncer, diabetes mellitus e infertilidad, entre otras.
Beneficios de la coenzima Q10: fertilidad y embarazo
Las posibilidades de embarazo disminuyen a medida que aumenta la edad. Esta situación puede estar motivada por diversidad de causas, pero el problema más común es la disminución en la calidad de los óvulos y espermatozoides.
El proceso para iniciar el desarrollo del embrión, una vez producida la fecundación, requiere una gran cantidad de energía, que es suministrada por las mitocondrias dentro de las células. En este sentido, los óvulos y espermatozoides tienen más mitocondrias que cualquier otra célula del cuerpo. En todo el proceso la coenzima Q10 juega un importante papel.
Algunos estudios muestran que la suplementación de coenzima Q10 en mujeres en tratamientos de fertilidad pueden mejorar la tasa de embarazo. De igual modo, la CoQ10 en hombres con infertilidad puede mejorar la motilidad de los espermatozoides, aumentar el número de espermatozoides viables y mejorar la penetración del espermatozoide en el óvulo.
En cualquier caso, la evidencia ofrecida por los estudios realizados hasta la fecha no permite ser concluyentes.
La coenzima Q10 y la preeclampsia
La preeclampsia es una complicación del embarazo que conduce a severas complicaciones a nivel cardiovascular que ponen en peligro las vidas de la madre y el bebé. Tiene un claro componente autoinmune y su principal síntoma es la hipertensión arterial.
En comparación con mujeres no embarazadas y embarazadas sanas, las pacientes con preeclampsia presentan concentraciones plasmáticas de coenzima Q10 significativamente más bajas.
En este sentido, el efecto del estrés oxidativo a nivel cardiovascular puede jugar un papel importante en la aparición de la preeclampsia, incrementando la producción de factores proinflamatorios y vasoconstrictores, los cuales determinan la degeneración vascular y la aparición de hipertensión arterial y proteinuria.
De este modo, la suplementación exógena de CoQ10 en pacientes con alto riesgo de desarrollar esta patología podría tener un efecto preventivo (disminuyendo a la mitad las posibilidades de padecerla).
La coenzima Q10 en la prevención de complicaciones en el embarazo
Existen estudios que han mostrado la efectividad de la CoQ10 en prevenir otras complicaciones, además de la preeclampsia.
Unos niveles adecuados del antioxidante han mostrado la capacidad de mejorar diferentes aspectos de la salud de la mujer embarazada, así como la del bebé.
Algunos de los beneficios asociados al CoQ10 incluyen:
Resultado final del embarazo . Existe una correlación positiva entre los niveles plasmáticos de coenzima Q10 y los resultados finales del embarazo. En los casos en los que el estrés oxidativo puede ocasionar ciertas complicaciones, los niveles de CoQ10 aparecen aumentados, como en la restricción del crecimiento intrauterino y la hipoxia intrauterina, entre otras situaciones.
Diabetes gestacional . En pacientes con diabetes del embarazo se observan incrementos en la concentración plasmática de CoQ10, lo que podría señalar un papel compensatorio para contrarrestar el efecto de estrés oxidativo causado por la hiperglucemia y la resistencia a la insulina.
Estilo de vida saludable durante el embarazo . Unos hábitos saludables que incluyan la realización regular de ejercicio físico, la evitación de tóxicos como el tabaco y el alcohol se relacionan con concentraciones plasmáticas elevadas de antioxidantes, incluyendo la coenzima Q10.
Reducción de marcadores proinflamatorios . La CoQ10 es capaz de reducir los niveles de algunos marcadores proinflamatorios que están asociados a la pérdida del embarazo.
Reducción de gestaciones anormales . Como se considera que la disfunción mitocondrial y el daño del material genético producidos por el estrés oxidativo pueden jugar un importante rol en la aparición de problemas en el embarazo, es posible que la coenzima Q10 contribuya a la prevención de los procesos degenerativos gracias a su cualidad antioxidante.
En cualquier caso, la relación entre unos niveles elevados de CoQ10 durante el embarazo y un mejor pronóstico y evolución del mismo parece claro. Sin embargo, no existen evidencias sólidas sobre la efectividad de su suplementación.
Contenidos sobre enfermedades
Somos conscientes de que los conceptos médicos pueden resultar confusos si no te los explican con claridad. Para evitar esto, le hemos pedido a nuestros profesionales que preparen estas fichas en donde podrás acceder a información relevante sobre enfermedades que te ayudará a entender de una manera sencilla por qué se producen y por qué es necesario su tratamiento.
Ver todas las enfemedades 
Enfermedad
Ovarios Poliquísticos
Actualizado el día 15/09/2021
El síndrome del ovario poliquístico es la patología endocrina más frecuente en mujeres en edad fértil, aunque permanece infradiagnosticado. Se trata de un síndrome complejo y heterogéneo en cuanto a su presentación, evolución y consecuencias a corto y largo plazo.
¿Qué es el síndrome de ovarios poliquísticos?
El síndrome de ovarios poliquísticos determina la aparición de quistes (pequeñas cavidades llenas de líquido) en los ovarios, debido a una producción excesiva de hormonas masculinas.
El síndrome de ovarios poliquísticos agrupa una colección de signos y síntomas que incluye, además de la producción de hormonas masculinas por encima de lo normal, disfunción ovulatoria con prolongación, disminución de la frecuencia o ausencia de los períodos menstruales con escasa liberación de óvulos y aparición de quistes en los ovarios .
El signo clínico más característico determinado por el hiperandrogenismo es el hirsutismo o exceso de vello siguiendo un patrón masculino. También puede aparecer acné y alopecia.
Esta alteración endocrina afecta aproximadamente a 1 de cada 10 mujeres en edad reproductiva y está relacionado directamente con la disminución de la fertilidad.
Causas del síndrome de ovarios poliquísticos
Como en la mayoría de los síndromes, no existe una única causa para los ovarios poliquísticos. Sí existe una disfunción neuroendocrina con un exceso de hormona luteinizante, que es la encargada de regular la secreción de testosterona, impidiendo a los óvulos madurar lo suficiente y que, en lugar de ser expulsados de los ovarios durante la ovulación, permanezcan en el interior formando los quistes.
También existe un trastorno metabólico con resistencia a la insulina e hiperinsulinemia, lo que también estimula la producción de andrógenos.
Otros factores que se relacionan con la enfermedad y que pueden influir en el desarreglo hormonal son la obesidad , una alimentación deficiente y una vida sedentaria, entre otros.
Finalmente, el síndrome de ovario poliquístico puede tener causas genéticas, por alteración o mutación de genes, lo que determina que sea una patología altamente hereditaria.
Síntomas
Los síntomas del síndrome de ovarios poliquísticos pueden incluir:
Menstruaciones irregulares . Es común la oligomenorrea (disminución hasta 6-8 reglas anuales) o amenorrea (ausencia de regla durante varios periodos menstruales seguidos). Estos ciclos menstruales irregulares se asocian con una disminución en la ovulación y mayores tasas de infertilidad.
Hirsutismo y síntomas virilizantes . El exceso de hormonas masculinas (hiperandrogenismo) determina la aparición de síntomas virilizantes, como el aumento de vello en diferentes zonas como la cara, muslos, abdomen, espalda y antebrazos. Otros síntomas incluyen clitoromegalia, alopecia, voz grave, hipotrofia mamaria y aumento de la masa muscular.
Acné y piel grasa.
La resistencia a la insulina puede conducir a la aparición de diabetes, hipertensión arterial, aumento del colesterol y obesidad, entre otros.
Aumento de peso .
Dolor pélvico .
Manchas de color marrón oscuro en la piel.
Pruebas y exámenes para el diagnóstico del síndrome de ovarios poliquísticos
En un tercio de las mujeres pasan más de 2 años hasta que se les diagnostica la enfermedad. De ahí que se deba poner el foco en dos cuestiones específicas para identificar a los pacientes con mayor riesgo: mujeres con exceso de vello corporal o facial con un patrón masculino y mujeres con un historial de irregularidad menstrual u oligoamenorrea.
Las pruebas complementarias y exámenes para el diagnóstico del síndrome de ovarios poliquísticos deben incluir:
Examen físico.
Examen pélvico.
Análisis de sangre completo que incluya determinaciones hormonales, en especial, las hormonas masculinas.
Ecografía ginecológica de alta resolución.
Tratamiento del síndrome de ovario poliquístico
El síndrome de ovario poliquístico no tiene cura, pero sí se pueden establecer metas terapéuticas para diferentes aspectos de la enfermedad.
Tratamiento no farmacológico
Los cambios en el estilo de vida pueden suponer una mejora considerable de los diferentes parámetros alterados por la enfermedad. La pérdida de peso, una alimentación sana y realizar ejercicio físico de manera regular puede favorecer la regulación hormonal, mejorar la sensibilidad a la insulina, aumentar la fertilidad y hacer que mejoren todos los síntomas.
Tratamiento farmacológico
Los anticonceptivos hormonales pueden ser adecuados para regular la producción de hormonas y de esta manera normalizar el ciclo menstrual. También pueden incidir positivamente sobre el acné y el crecimiento del vello corporal.
El tratamiento hormonal como los antiandrógenos que regulan el nivel de las hormonas masculinas mejoran el acné y el vello corporal.
Por otro lado, el uso de antidiabéticos orales como la metformina puede ayudar a regular los niveles de insulina y, por tanto, a regular los ciclos menstruales.
Por último, se pueden utilizar inductores de la ovulación en el caso de pacientes que deseen quedarse embarazadas.
Factores desencadenantes del síndrome del ovario poliquístico
Como en la mayoría de síndromes, no existe un único desencadenante del síndrome del ovario poliquístico.
Por el contrario, existen diversos factores que pueden determinar un mayor riesgo de que aparezca la patología.
Factores de riesgo de los ovarios poliquísticos
Los factores de riesgo del síndrome de ovarios poliquísticos pueden incluir una alimentación deficiente, el exceso de peso y la obesidad y una vida sedentaria. También puede haber factores genéticos que hacen que la enfermedad sea hereditaria en base a determinados cambios o mutaciones genéticas.
Complicaciones
Las mujeres que padecen el síndrome de ovario poliquístico pueden presentar una serie de complicaciones asociadas, entre ellas:
Síndrome metabólico.
Diabetes tipo 2 y obesidad.
Hipertensión arterial.
Enfermedades cardiovasculares.
Trastornos menstruales, infertilidad y abortos.
Crecimiento excesivo de vello.
Apnea obstructiva del sueño.
Cáncer de endometrio.
Depresión y ansiedad.
Prevención
Existe una serie de recomendaciones que pueden ayudar a reducir el riesgo de padecer la enfermedad y, en todo caso, atenuar los síntomas y regular los ciclos menstruales. Además del tratamiento médico, es importante conseguir una buena calidad de vida eliminando el estrés, realizar ejercicio físico de manera regular, mantener una dieta saludable y evitar el exceso de peso y la obesidad.
Especialidades para el síndrome del ovario poliquístico
El síndrome de ovarios poliquísticos es tratado por los médicos especialistas en Endocrinología y en Ginecología.
¿Necesitas atención presencial con un especialista en ginecología? En Savia reserva una consulta con los mejores profesionales sin esperas.
Preguntas frecuentes
¿Cuáles son los síntomas de quistes en los ovarios?
Los síntomas de quistes en los ovarios son dolor en la parte baja del abdomen, hinchazón y sensación de plenitud abdominal, dolor durante las relaciones sexuales o dolor durante la defecación, entre otros.
¿Qué riesgos implica el ovario poliquístico?
El síndrome de ovario poliquístico aumenta el riesgo de infertilidad, cáncer de endometrio, metabolismo anormal de la glucosa y dislipidemia.
¿Qué produce quistes en los ovarios?
Los quistes de ovarios suelen tener un componente genético, pero también se producen por una alteración hormonal, endometriosis, embarazo, infecciones pélvicas o el síndrome de ovarios poliquísticos.
¿Cuál es el tamaño normal de un quiste de ovario?
Un quiste de ovario suele medir menos de 5 cm. En el síndrome de ovarios poliquísticos suelen aparecer al menos 12 folículos que suelen medir de 2 a 10 mm.
¿Qué son los folículos de los ovarios?
Los folículos son estructuras anatómico-funcionales que forman parte del ovario y que albergan en sus paredes internas al ovocito y a otras células productoras de estrógenos.
¿Qué es un quiste funcional?
En los ovarios se producen folículos que son estructuras funcionales que producen hormonas y liberan el óvulo durante la ovulación. Cuando un folículo normal continúa creciendo, se denomina quiste funcional. No suelen causar síntomas y desaparecen por sí solos.
¿Cuánto tiempo tarda en quitarse un quiste de ovario?
Por norma general, un quiste de ovario se rompe en el momento de la ovulación, pero algunos resisten y pueden tardar hasta 3 meses en desaparecer.
Bibliografía
Sociedad Española de Ginecología y Obstetricia. Estudio y tratamiento de la anovulación en el síndrome de ovarios poliquísticos. Prog Obstet Ginecol 2017; 60(5): 505-516 .
Azziz R. Polycystic Ovary Syndrome. Obstet Gynecol 2018; 132(2): 321-336. doi: 10.1097/AOG.0000000000002698 .
McCartney CR, Marshall JC. Polycystic Ovary Syndrome. N Engl J Med. 2016 Jul 7;375(1):54-64. doi: 10.1056/NEJMcp1514916 .

Enfermedad
Síndrome premenstrual
¿Qué es el síndrome premenstrual?
El síndrome premenstrual (SPM) es un conjunto de síntomas y cambios físicos y psíquicos, bien definidos, que experimentan las mujeres días antes de la menstruación (entre el día 14 y 28). Esto sucede mes tras mes y puede afectar al 90 % de las mujeres, y con mayor frecuencia entre los 20 y 30 años de edad.
La intensidad de los síntomas puede variar de unas mujeres a otras y pueden ser simples molestias o ser tan agudos que el malestar que ocasionan pueden interferir e incluso incapacitar en las actividades diarias o laborales.
El síndrome premenstrual termina cuando empieza la regla .
Tipos de síndrome premenstrual
No existen tipos de síndrome premenstrual.
Causas de un síndrome premenstrual
No se sabe con exactitud cuales son las causas del síndrome premenstrual , pero podría estar relacionado con un desequilibrio hormonal de estrógenos y progesterona. Esto produce una retención de líquidos y sodio que provocaría un aumento de peso y edema generalizado dando lugar a los síntomas premenstruales.
También se desconocen las causas por las que unas mujeres son más sensibles que otras a estos cambios hormonales.
Síntomas de un síndrome premenstrual
Los síntomas del síndrome premenstrual pueden ser:
Síntomas físicos:
Hinchazón de abdomen
Edemas generalizados por retención de líquidos
Tensión e inflamación de las mamas
Diarrea o estreñimiento
Calambres
Dolor de cabeza y espalda
Intolerancia a la luz y al ruido
Acné
Síntomas psicológicos o emocionales:
Cambios repentinos del estado de ánimo
Irritabilidad
Cansancio
Ansiedad
Tristeza o depresión
Cambios de apetito como la necesidad de comer dulce
Trastornos del sueño
Pérdida de interés en el sexo
Pérdida de concentración
Tratamiento de un síndrome premenstrual
El tratamiento del síndrome premenstrual puede ser:
Tratamiento médico: para aliviar los síntomas como los antiinflamatorios, analgésicos, diuréticos, antidepresivos o anticonceptivos hormonales, entre otros.
Remedios caseros: ayudan a paliar los síntomas como la actividad física diaria, evitar la sal, cafeína, alcohol y azúcar los días previos al ciclo, no fumar, controlar el estrés, descansar y dormir más horas, o llevar una dieta saludable con más vitaminas y minerales durante esos días previos.
Pruebas complementarias de un síndrome premenstrual
No hay pruebas específicas para diagnosticar el síndrome premenstrual , basta con la descripción de los síntomas, en qué momento aparecen y si se repiten durante al menos tres ciclos menstruales para poder diagnosticar el síndrome premenstrual.
Factores desencadenantes de un síndrome premenstrual
El factor desencadenante se debe a la disminución de los niveles de estrógeno y progesterona después de la ovulación. Cuando comienza la menstruación, los niveles de estas hormonas comienzan a subir de nuevo. S e desconoce porqué unas mujeres son más sensibles que a otras .
Factores de riesgo de un síndrome premenstrual
No hay factores de riesgo.
Complicaciones de un síndrome premenstrual
Las posibles complicaciones de un síndrome premenstrual pueden ser la incapacidad del desarrollo de las actividades diarias y laborales, pensamientos suicidas e incluso suicidio en las mujeres con depresión, por eso es importante ajustar el tratamiento esos días puntuales .
Prevención de un síndrome premenstrual
No existe prevención para evitar el síndrome premenstrual.
Especialidades a las que pertenece
El síndrome premenstrual es tratado por el médico de Atención Primaria , que trata los síntomas y recomienda pautas para aliviarlos, pero cuando el síndrome premenstrual afecta de manera más intensa o incapacita el ritmo de vida diario es el Ginecólogo (médico del aparato reproductor femenino) quien hace un seguimiento y tratamiento del síndrome premenstrual.
Preguntas frecuentes
¿Cuándo aparecen los primeros síntomas del embarazo?
Los primeros síntomas de embarazo es la ausencia de regla, senos hinchados, cansancio y somnolencia, orinar más a menudo, náuseas y vómitos, hinchazón abdominal y dolor pélvico, entre otros.
¿Cuándo se presenta el síndrome premenstrual?
El síndrome premenstrual se presenta cuando aparecen una serie de síntomas concretos días antes de la regla, durante al menos 3 meses.
¿Cuánto es lo máximo que dura el periodo menstrual?
El periodo menstrual puede durar entre 25 y 30 días, incluso alargarse a 35 días.
¿Cuáles son los trastornos menstruales?
Los trastornos menstruales pueden ser náuseas, vómitos, dolor abdominal tipo retortijón, dismenorrea (dolor menstrual), dolor de espalda, diarrea o dolor de cabeza, entre otros.
¿Cuánto tiempo pueden durar los síntomas premenstruales?
Los síntomas pre menstruales duran unos días antes de la aparición de la regla. Suelen estar presentes en los años entre la adolescencia y la vida adulta, es decir, entre los 20 y 30 años.

Enfermedad
Mioma Uterino
¿Qué es el mioma uterino?
El mioma o fibroma uterino, es un tipo de tumoración benigna en las paredes del útero y está formado por un incremento en forma de nódulo de las fibras musculares del útero .
Los miomas varían en número, tamaño y localización. Afecta a mujeres en edad fértil y su origen es desconocido , pero su crecimiento está muy influenciado por las hormonas femeninas.
Tipos de mioma uterino
Los miomas uterinos pueden ser:
Mioma subseroso: tiene una afectación en mujeres del 40% de miomas. No produce síntomas, pero si alcanza un tamaño grande puede producir molestias y dolor por compresión en órganos próximos.
Miomas intramusculares: debido a que pueden alcanzar gran tamaño, puede aumentar, también, el tamaño del útero. Constituye el 55% de los miomas y se suelen localizar en la porción central del miometrio (capa muscular intermedia).
Mioma submucoso: son menos frecuentes, dan más sintomatología al producir mucho sangrado menstrual provocando, en muchos casos, que la mujer padezca anemia ferropénica (falta de hierro). Tiene una elevada probabilidad de malignizar.
Causas de un mioma uterino
No se conocen las causas del mioma de útero, pero se ha detectado una relación directa con las hormonas femeninas , condicionando el desarrollo y crecimiento de los miomas, por eso se da en mujeres en edad fértil (desde la pubertad hasta la menopausia). Transcurrido este tiempo, el tamaño disminuye debido a que los niveles de hormonas en sangre son menores.
Síntomas de un mioma uterino
Aproximadamente el 30% de las mujeres no tienen ningún síntoma y solo se descubre su presencia de un mioma uterino en las revisiones rutinarias con el ginecólogo .
La aparición de los síntomas depende de su tamaño, número de miomas y localización, siendo los más sintomáticos los que se localizan en la cavidad interna del útero.
Los síntomas pueden ser:
Cambios menstruales en su duración, frecuencia y cantidad de sangrado.
Dolor.
Anemia ferropénica debido al exceso de sangrado.
Aumento del perímetro del abdomen en caso de miomas grandes.
Aumento de peso.
Complicaciones en embarazo y parto.
Compresión de vejiga y recto en el caso de miomas de gran tamaño.
Necesidad de orinar con mayor frecuencia.
Lumbalgia y dolor en pelvis.
Infertilidad.
Aumento de probabilidades de aborto o parto prematuro.
Molestias en las relaciones sexuales.
Tratamiento de un mioma uterino
La mayoría de los miomas no requieren tratamiento por ser asintomáticos, y se tiende a tener una actitud conservadora.
En caso de presentar sintomatología, el tratamiento puede ser:
Tratamiento médico:
Fármacos (agonistas de la GnRH) que disminuyen el nivel de estrógenos en sangre y, con ello, el tamaño de los miomas, pero tiene efectos secundarios con sintomatología similar a una menopausia (sofocos, sudoración, sequedad vaginal, etc.) y, además, cuando se termina el tratamiento, el mioma vuelve a crecer.
Analgésicos para paliar el dolor.
Anticonceptivos, progestágenos y dispositivos uterinos para disminuir la hemorragia.
Hierro para tratar la anemia.
Tratamiento quirúrgico:
Se emplea dependiendo de tamaño y localización del mioma.
Se puede realizar por vía vaginal o laparoscópica (tubo flexible con una cámara e instrumentos quirúrgicos, que se introduce por unas incisiones realizadas en el abdomen). Pueden ser:
Miomectomía, en la que se extirpa sólo el mioma, se conserva el útero y se preserva la fertilidad.
Histerectomía, en la que se extirpa una parte o la totalidad del útero, dependiendo de la localización y tamaño del mioma. Se preserva la función hormonal por conservar los ovarios, pero no hay posibilidad de embarazo.
Radiología intervencionista: técnica guiada por rayos usada para embolizar los miomas, es decir, suspender el riego sanguíneo del mioma.
Pruebas complementarias de un mioma uterino
Algunas pruebas complementarias para el diagnóstico de un mioma uterino son:
Exploración física: para detectar el tamaño del útero, presencia de miomas y dolor a la palpación.
Ecografía: para localizar, medir y determinar cuántos miomas hay. Es la prueba diagnóstica más efectiva y fiable.
Histeroscopia: se introduce un tubo por la vagina para ver el interior del útero.
TAC pélvico: para observar la repercusión de los órganos vecinos.
Resonancia magnética: para visualizar posibles tumores.
Factores desencadenantes de un mioma uterino
Los factores desencadenantes de los miomas uterinos están causados por el aumento de estrógenos en sangre .
Factores de riesgo de un mioma uterino
Los factores de riesgo de un mioma pueden ser:
Mujeres de entre 35 y 45 años.
Mayor incidencia en la raza afroamericana.
Obesidad, hipertensión o diabetes.
Mujeres nulíparas (no han dado a luz).
Desarreglo hormonal (aumento de estrógenos).
Factor hereditario.
Complicaciones de un mioma uterino
Las complicaciones del mioma uterino pueden ser:
Compresión de vejiga y retención urinaria.
Edemas, varices y trombosis en las piernas, por compresión de venas pélvicas dificultando la circulación de retorno de las piernas.
Hemorragias abundantes.
Malignización del mioma, suele ocurrir en un 0,5 % de los miomas.
Prevención de los miomas
Los miomas no se pueden prevenir debido a que no se sabe con exactitud la causa .
Se pueden hacer unas recomendaciones como son las revisiones ordinarias con el ginecólogo, controlar el peso y llevar una dieta saludable junto con la práctica regular de ejercicio físico.
Especialidades a las que pertenece
La especialidad médica que trata los miomas es Ginecología y Obstetricia.
Preguntas frecuentes
¿Cuál es el tamaño de un mioma uterino?
Los miomas varían en tamaño, siendo los más pequeños de hasta 2 cm. y los grandes a partir de 6 cm .
¿Cuál es la diferencia entre un mioma y un fibroma?
Es lo mismo un mioma que un fibroma. También se pueden llamar leiomiomas .
¿Qué son miomas tipo 4?
Los miomas tipo 4 son miomas que ocupan un 50% del volumen del interior del útero .
¿Qué es un mioma en el endometrio?
Un mioma en el endometrio es aquel que crece hacia la cavidad uterina, es decir, hacia la mucosa que recubre la pared del útero .
¿Cuándo una mujer tiene miomas puede quedar embarazada?
Los miomas son compatibles con el embarazo, pero, en ocasiones, pueden aparecer complicaciones durante la gestación (abortos, partos prematuros o alteración en el desarrollo fetal), en el parto (hemorragias, mala presentación del feto o dificultad en las contracciones), y en el postparto (hemorragias).

Enfermedad
Vaginosis Bacteriana
¿Qué es la vaginosis bacteriana?
La vaginosis bacteriana es una infección vaginal por una bacteria , se produce por el desequilibrio entre la diferente flora bacteriana de la vagina, en la cual se produce un descenso de los lactobacilos y un aumento de la gardnerella (bacilo de la región genital). Es la infección bacteriana más frecuente entre los 14 y 40 años, y es una enfermedad leve.
Tipos de vaginosis
Los tipos de vaginosis serán diferentes según el germen que lo produce, así tenemos:
Vaginosis bacteriana : se produce por el germen conocido como Gardnerella vaginalis.
Vaginitis candidiasica o candidiasis vaginal : se debe a la infección por hongo Candida albicans , produce un flujo blanco y espeso.
Vaginitis tricomoniasis : se produce por el protozoo Trichomona vaginalis.
Vaginitis por clamidia : producida por la Chlamydia trachomatis
Causas de la vaginosis bacteriana
La causa de la vaginosis bacteriana es la infección por parte de la Gardnerella vaginalis.
Síntomas de vaginosis bacteriana
La vaginosis bacteriana en la mayoría de los casos es asintomática , o presenta síntomas muy leves. Cuando aparecen síntomas, estos son, la presencia de flujo delgado y abundante con olor a pescado , de un color que puede ser blancuzco, gris apagado o incluso verdoso, las pacientes también pueden presentar ciertas molestias como ardor al orinar o ligero picor, en algunos casos poco frecuentes puede dar dolor suprapúbico.
Tratamiento para la vaginosis bacteriana
El tratamiento de la vaginosis bacteriana va a ser tratamiento antibiótico , que puede ser de uso oral, en forma de óvulos vaginales, o geles. Los antibióticos que se suelen usar son el Metronidazol o Clindamicina .
Pruebas complementarias del tratamiento de vaginosis bacteriana
La prueba complementaria será un exudado vaginal , aunque también se puede encontrar mediante citología en aquellas personas que no presentan sintomatología y por tanto no consultan por este problema.
Factores desencadenantes de la vaginosis bacteriana
No se conocen las causas que desencadenan las vaginosis bacterianas, si bien se conoce que las personas con relaciones sexuales con múltiples parejas pueden sufrir esta infección. No se encuentran casos de vaginosis bacteriana en personas que no tienen relaciones sexuales.
Factores de riesgo de la vaginosis bacteriana
Son factores de riesgo las duchas vaginales, el uso de productos de higiene femenina muy perfumados o que puedan ser irritantes, y el uso de ropa ajustada y que no transpire.
Complicaciones de la vaginosis bacteriana
La vaginosis bacteriana es una infección local que no presenta complicaciones.
Prevención de la vaginosis bacteriana
Evitar las duchas vaginales.
Evitar el uso de jabones abrasivos.
Usar preservativo para evitar contagios.
Ropa interior de algodón.
Especialidades a las que pertenece la vaginosis bacteriana
La vaginosis bacteriana será tratada en primer lugar por el médico de familia, solo en casos que sean recidivantes puede ser necesario el tratamiento por parte del ginecólogo.
Preguntas frecuentes
¿Qué es la candidiasis bacteriana?
No existe la candidiasis bacteriana. La candidiasis es un infección producida por un hongo conocido como Cándida albicans , que, al reproducirse por alteraciones locales en mayor cuantía de lo normal, da lugar a una infección que se caracteriza por picor intenso , asociado a producción de flujo blanquecino y espeso. Las bacterias son un tipo de células diferentes que producen otras infecciones como la vaginosis bacteriana.
¿Qué es la vulvovaginitis?
La vulvovaginitis es la inflamación tanto de la vulva como de la vagina, la paciente presenta síntomas internos de molestias vaginales, asociados a picor externo y, en muchos casos, ardor al orinar, ya que el paso de la orina da lugar a un irritación local de la vulva. Precisará tratamiento interno de la vagina y externo de la vulva para mejorar los síntomas. Puede ser producida por bacterias y hongos.
¿Hay algún remedio casero para aligerar los síntomas de la vaginosis bacteriana?
Para tratar la vaginosis bacteriana son necesarias medidas que cambien el PH vaginal, haciéndolo más ácido. Así, al añadir vinagre de manzana al agua de lavarse puede ser eficaz, el consumo y aplicación de yogur natural puede ser otro tratamiento, añadir al agua de lavarse el aceite del árbol de té puede ser también cambiar el PH vaginal, impidiendo las infecciones de repetición. Evitar el uso exagerado de productos de higiene femenina que pueden ser irritantes y cambiar el PH por sí mismo.
¿Qué es la enfermedad Gardnerella?
La Gardnerella es una bacteria conocida como Gardnerella vaginalis , que produce una infección en la vagina conocida como vaginosis bacteriana, que se debe a una alteración de la flora vaginal donde disminuyen los Lactobacillus y prolifera la Gardnerella , es la principal causa de alteración vaginal aunque hasta el 50 de las personas que la presentan no tiene síntomas.
¿Qué es una célula clave?
La célula clave es una célula pavimentosa de la vagina, que se encuentra rodeada de bacterias, en general Gardenella vaginaleis , las baterias se hayan pegadas a la pared celular y son típicas de la infección por esta bacteria.
Artículos por especialidad
CertificadosAlergologíaAnatomía PatológicaAnestesiología y ReanimaciónAngiología y Cirugía VascularAnálisis ClínicosAparato DigestivoCardiologíaChequeosCirugía General y del Aparato DigestivoCirugía General y del Aparato Digestivo PediátricaCirugía Oral y MaxilofacialCirugía Ortopédica y TraumatologíaCirugía Ortopédica y Traumatología PediátricaCirugía PediátricaCirugía Plástica y ReparadoraCirugía TorácicaClínica u HospitalDermatología Médico-Quirúrgica y VenereologíaEndocrinología y NutriciónEnfermeríaFisioterapiaGenética HumanaGinecología y ObstetriciaInmunologíaLogopedia y FoniatríaMedicina Estética y Unidad LaserMedicina Física y RehabilitaciónMedicina GeneralMedicina InternaMedicina NuclearMedicina del DeporteNefrologíaNeumologíaNeurocirugíaNeurofisiología ClínicaNeurologíaNutrición y DietéticaOdontología y EstomatologíaOftalmologíaOncología MédicaOncología RadioterápicaOsteopatíaOtorrinolaringologíaPediatríaPodologíaPoliclínicasPreparación al PartoPsicologíaPsiquiatríaRadiodiagnóstico-Diagnóstico Por ImagenReumatologíaTricologíaUnidad del DolorUrgencias HospitalariasUrología
Todas las especialidades ¿Puedo confiar en Savia?
Todos nuestros servicios están creados con la intención de acercar la atención médica y sanitaria de calidad a todos los trabajadores y sus familias, siempre con la garantía de MAPFRE. Queremos establecer una relación de total confianza.