Artículos Especializados
Artículo especializado
CIR en el embarazo: crecimiento intrauterino restringido
El crecimiento fetal durante la gestación está regulado por factores maternos, fetales y placentarios. Todos ellos en coordinación harán que se asegure la llegada de los nutrientes maternos al feto en la forma y proporción adecuadas, consiguiéndose un crecimiento fetal optimo .
El desarrollo normal de estos condicionantes permite al feto obtener el crecimiento que genéticamente está determinado, desarrollando todo su potencial intrauterino. Por el contrario, cuando existen alteraciones en alguno de los factores, se pueden producir retrasos y restricciones en el crecimiento fetal dentro del útero. Es lo que se conoce como crecimiento intrauterino restringido (CIR) .
¿Qué es el CIR o crecimiento intrauterino restringido?
El crecimiento intrauterino restringido (CIR) es una condición en la que el crecimiento del feto dentro del útero se ralentiza o se detiene.
Se trata de un problema relativamente común que puede llegar a afectar a cerca del 10% de los embarazos en diferente grado, manteniéndose como unos de los grandes retos en la práctica obstétrica actual.
La consecuencia principal del CIR es que el bebé no alcance el tamaño previsto, presentando por ello mayor riesgo de padecer complicaciones en el momento de nacer y también tras el parto. Por ejemplo, muerte intrauterina, prematuridad y peor desarrollo neurológico, entre otras.
¿Cuándo se detecta un CIR?
El grado de desarrollo del feto durante el embarazo, así como la posible situación de CIR es detectada a partir de los controles ecográficos periódicos a los que se someten las embarazadas para evaluar el crecimiento fetal.
Cuando el crecimiento fetal durante toda la gestación es inferior a lo esperado, se considera que es un feto con CIR de aparición precoz. En este caso suele deberse a anomalías congénitas o infecciones prenatales.
Por el contrario, cuando el crecimiento es normal y en el tercer trimestre de gestación se ralentiza o se detiene, se tratará de un CIR de aparición tardía, en cuyas causas estarán involucrados con toda probabilidad los factores maternos o placentarios. La causa más común es una insuficiencia placentaria causada por hipertensión materna. El pronóstico en estos casos es bueno.
De este modo, los problemas en el funcionamiento de la placenta suele ser la causa más común. No en vano se trata del tejido encargado de transportar el alimento y el oxígeno al feto.
En cuanto a los factores maternos de riesgo podemos mencionar:
La edad de la madre (menor de 16 o mayor de 35 años).
El bajo nivel socioeconómico.
Haber transcurrido menos de 6 meses entre dos gestaciones.
Consumo de tabaco, alcohol y drogas.
El tratamiento con determinados fármacos por parte de la madre.
Patologías maternas como asma, hipertensión arterial, diabetes mellitus, patologías renales, etc.
Infecciones y parasitosis en la madre (toxoplasmosis, citomegalovirus, malaria, VIH, etc.).
Entre los factores fetales se pueden encontrar:
Anomalías cromosómicas, como el síndrome de Down y otros síndromes genéticos complejos.
Anomalías congénitas con afectación cardíaca, neurológica, etc.
Gestaciones múltiples.
Enfermedades metabólicas.
¿Qué es un CIR tipo 1?
El CIR de tipo 1 tiene lugar durante el primer trimestre del embarazo, es decir, se trata de CIR precoz. En este caso, la disminución en el crecimiento es proporcional, por lo que se produce un menor crecimiento, pero simétrico y armónico en el que todas las medidas ecográficas están disminuidas. Por motivos diversos (incluyendo la genética) el feto recibe menos nutrientes de lo necesario, por lo que su tamaño es más reducido. La insuficiencia placentaria que se produce es severa, la prematuridad mayor y el pronóstico más grave.
Por el contrario, en el CIR tipo 2 o asimétrico se produce a partir de la semana 34 del embarazo, es tardío y es el más frecuente. Generalmente, se asocia a hipertensión materna durante el embarazo. En este caso el crecimiento es desigual, mostrándose una cabeza, los huesos largos y el abdomen más grandes y desproporcionados que el resto del cuerpo. En el tipo 2, los nutrientes se dirigen prioritariamente al cerebro y las partes más importantes del cuerpo, por lo que se desarrollan en mayor medida.
¿Cómo se diagnóstica el crecimiento intrauterino retardado?
Se considera que el peso del recién nacido es bajo cuando es inferior a 2,5 kg, independientemente de si ha sido pretérmino y de su edad gestacional. De igual modo, un bebé pequeño para su edad gestacional es aquel que está por debajo del percentil 10, en las curvas de percentiles, situándose el crecimiento normal en 50, pero sin que concurran trastornos en el desarrollo.
Por el contrario, el crecimiento intrauterino restringido o retardado se produce cuando se ha producido un crecimiento anormal dentro del útero materno. En este caso, el embrión no ha sido capaz de desarrollarse plenamente dentro del vientre materno.
La prueba principal para establecer el diagnóstico de CIR es la ecografía. Las medidas del feto permiten estimar su peso y establecer en qué percentil de crecimiento se encuentra, teniendo en cuenta otros factores como el sexo, origen étnico, etc.
El estudio de crecimiento fetal se complementa con el estudio Doppler, que permite evaluar el funcionamiento de la placenta y el grado de oxigenación del feto.
Cuando se ha establecido el diagnóstico y en función del momento de aparición del retraso de crecimiento, se pueden realizar otras pruebas para valorar las posibles causas:
Indicada en casos graves y antes de la semana 26.
Analítica para el estudio de posibles infecciones y parasitosis.
Analítica para identificar alteraciones compatibles con preeclampsia.
Seguimiento periódico ecográfico del crecimiento fetal.
Para hacer el seguimiento del desarrollo fetal la herramienta más útil es la ecografía obstétrica .
¿Qué pasa cuando el bebé deja de crecer en el vientre?
Los bebés que han experimentado un crecimiento intrauterino restringido presentan un mayor riesgo de sufrir determinados problemas de salud. Además de requerir mayores cuidados y un mayor periodo de hospitalización tras el nacimiento, pueden presentar:
Problemas respiratorios y para alimentarse.
Dificultad para mantener una temperatura estable en el cuerpo.
Recuento sanguíneo anormal.
Hipoglucemia
Menor capacidad inmunitaria.
Problemas neurológicos.
Además, también existirá un mayor riesgo para la salud en etapas posteriores de la vida, incluso en la edad adulta, incluyendo enfermedades cardiovasculares y diabetes, entre otras.
El correcto seguimiento del embarazo permitirá un diagnóstico precoz y oportuno, pudiendo tomar medidas para disminuir las complicaciones presentes y futuras en estos casos.
Artículo especializado
Lunares en bebés: ¿Cuándo empiezan a salir?
Los lunares de los recién nacidos son lunares o nevus congénitos, un tipo de lesión de la piel que está presente ya en el nacimiento. Es importante establecer un control sobre su evolución para evitar complicaciones, especialmente la aparición de un melanoma.
¿Cuándo empiezan a salir los lunares a los bebés?
Los lunares son una proliferación benigna de melanocitos, las células encargadas de producir la melanina que da color a la piel. Gran cantidad de lunares se desarrollan durante la infancia y adolescencia y su número se estabiliza hacia los 25 años de edad. Aproximadamente el 1% de los recién nacidos presentan algún lunar congénito. La gran mayoría no suponen ningún riesgo, si bien en el caso del nevus melanocítico congénito gigante y los nevus clínicamente atípicos pueden requerir extirpación.
Además de los lunares congénitos, presentes al nacer, también existen los lunares adquiridos, que suelen aparecen después de los 6-12 meses de edad y aumentan de tamaño con el crecimiento. Suelen ser de un tamaño inferior a 5 mm de diámetro, son simétricos y muestran una coloración uniforme. Su número es muy variable entre individuos y pueden aparecer en cualquier zona de la piel.
¿Cuándo preocuparse por lunares en los bebés?
Como se ha dicho, la gran mayoría de los lunares congénitos no suponen un problema para el bebé, pero es preciso hacer un seguimiento de los mismos. Es normal que aumenten de tamaño a medida que el bebé vaya creciendo. Debe ser el Dermatólogo el que determine el grado de seguimiento y las precauciones que se deberán tomar. Por lo general, se tendrá en cuenta el tipo de piel, la cantidad y tipo de nevus, el momento de su aparición y los antecedentes familiares, tanto de lunares como de cáncer de piel.
En el caso del melanoma, los factores de riesgo en el niño incluyen:
La presencia de nevus melanocítico congénito.
Ser portador de un número importante de nevus y nevus atípicos.
Es importante identificar las lesiones melanocíticas con un riesgo aumentado de malignización que requieran un diagnóstico y tratamiento precoz. Aunque el melanoma maligno es un tumor infrecuente en el niño, en el adulto es un cáncer que está aumentando su prevalencia y también en adolescentes y jóvenes de 14 a 20 años.
Causas y riesgos de los lunares en bebés
Los lunares congénitos están presentes en aproximadamente el 1% de los recién nacidos, incluyendo cualquier lunar que aparece durante las dos primeras semanas de vida. Estos se pueden clasificar según su tamaño en:
Pequeños (menores de 2 cm). Se suele recomendar la extirpación en lesiones muy oscuras o de aspecto multinodular en las cuales la detección de una transformación a melanoma sería complicada, al igual que los localizados en zonas de difícil seguimiento, como el cuero cabelludo, plantas de los pies o la región genital.
Es recomendable diferir la extirpación hasta una edad del niño que le permita colaborar en la intervención con anestesia local.
Intermedios (de 2 a 20 cm). Pueden ser precursores de melanoma y se debe considerar su extirpación preventiva para disminuir el riesgo. Deben ser evaluados siempre de forma individualizada.
Gigantes (más de 20 cm). Presenta una pigmentación irregular con un aspecto infiltrado y multinodular en ocasiones con un vello grueso y tosco. Se estima que el riesgo acumulativo probable en toda la vida de transformación maligna es del 5 al 15 %. Este riesgo parece ser mayor en los primeros años de vida (de 3 a 5 años). Se suele recomendar su extirpación temprana.
Factores de riesgo para la aparición y multiplicación de lunares
Más allá de los lunares congénitos, uno de los factores claramente relacionados con un mayor número de nevus es la exposición al sol. Esta exposición aumentada, en combinación con las características personales de cada individuo, como los fototipos de piel I y II (ojos claros, piel clara, pelo rubio o pelirrojo) influye en un mayor número de nevus y en el riesgo aumentado de transformación maligna.
En el melanoma, el riesgo se relaciona principalmente con la sensibilidad solar y un exceso de exposición solar y quemaduras solares durante la infancia. Por ello, es imprescindible una protección contra el sol, sobre todo durante la infancia y adolescencia, especialmente en casos de piel clara con múltiples lunares o si estos son atípicos. Es importante utilizar cremas que protejan contra la radiación ultravioleta A (UVA) y B (UVB). Además de limitar la exposición y las cremas protectoras, es recomendable el uso de prendas de vestir y gafas de sol.
Por otro lado, la depresión inmunitaria, independientemente de su causa, también se relaciona con un mayor número de lunares.
Por último, también puede incidir un factor genético en la probabilidad de una transmisión familiar. Es el caso del síndrome familiar, asociando un gran número de nevus de características atípicas con un riesgo importante de aparición de melanoma.
Tratamiento
Prácticamente todas las personas son portadoras de algún lunar, siendo la mayor parte de estas lesiones inocuas. La transformación maligna de un nevus melanocítico congénito es infrecuente, excepto en los nevus congénitos gigantes.
En cualquier caso, el objetivo principal del tratamiento es reducir el riesgo de desarrollar un cáncer de piel y, en un segundo término, mejorar el aspecto estético del niño, si este se ha visto comprometido.
En los lunares pequeños se recomienda un control periódico de los cambios en el color, el tamaño o en el aspecto de sus bordes.
En los lunares medianos se recomienda una biopsia para analizar su estructura interna. Cuando son superficiales el riesgo de malignización es bajo. Sin embargo, cuando son profundos, existe un mayor riesgo y se recomienda su extirpación mediate cirugía.
Finalmente, en los nevus gigantes el tratamiento es más complejo por el área afectada. Se deben extirpar precozmente y reparar la zona de la piel afectada mediante injertos.

Artículo especializado
Siringomas: ¿Por qué salen y cómo se tratan?
Existen muchas afecciones benignas de la piel que no ponen en riesgo la salud, pero que pueden tener un elevado impacto estético. Entre ellas se encuentran los siringomas.
¿Qué son los siringomas?
Los siringomas o hidradenomas eruptivos son pequeños tumores benignos de la piel procedentes de la porción intradérmica de los conductos de las glándulas sudoríparas. Suelen afectar a mujeres jóvenes.
Se caracterizan por la aparición de pápulas de forma redondeada, firmes al tacto, habitualmente de 1 a 3 mm de diámetro, del mismo color de la piel o un color ligeramente más claro (amarillento o rosado). Pueden aparecer en cualquier zona de la piel, aunque suelen localizarse en diferentes partes del cuerpo, especialmente en los párpados (habitualmente en el párpado inferior o justo debajo de él). También pueden afectar al cuello, tronco, abdomen y los genitales. Menos común es su aparición en mejillas, frente, pecho, axilas, palmas de las manos y en el cuero cabelludo.
Los siringomas que aparecen en torno a los ojos suelen presentarse de forma simétrica en ambos ojos.
Las erupciones pueden aparecer en un número variable y suelen agruparse, formando placas.
Esta afección suele ser asintomática, si bien en ocasiones puede provocar picor.
La consecuencia más importante de los siringomas es su afectación estética.
Causas de los siringomas
Las causas que están detrás de la aparición de siringomas no son del todo conocidas. Sin embargo, existe una clara predisposición a padecerlas en determinadas personas.
Debido a que los siringomas en la zona vulvar son más frecuentes en adolescentes, mujeres en edad reproductiva y embarazadas, se considera como posible agente causal un componente de origen hormonal.
En cuanto a la causa hereditaria, se ha descrito un patrón de herencia autosómica dominante.
Otras teorías causales apuntan a procesos inflamatorios crónicos de la piel con implicación autoinmune.
Además, puede haber diferentes factores que pueden contribuir a un mayor riesgo de padecer siringomas:
Factores hereditarios.
Producción excesiva de glándulas sudoríparas.
Fototipos oscuros de piel.
Sexo femenino.
Adolescencia.
Patologías como la diabetes y el hipertiroidismo.
Síndromes y patologías genéticas, incluyendo el Síndrome de Down. Otras asociaciones posibles incluyen el síndrome de Ehlers-Danlos, síndrome de Marfan, síndrome de Nicolau and Balus, síndrome de Klippel Feil, enfermedad de Fabry, nevo de Spitz, liquen amiloide, prurigo nodular, quiste eruptivo velloso, vitíligo y eccema.
Tipos de siringomas
Los siringomas, dependiendo de las características morfológicas y la zona en la que aparecen, se pueden diferenciar en:
Siringoma palpebral. Se trata del tipo más frecuente y aparece en la zona de los párpados. Suele iniciarse tras la pubertad. Las lesiones son del tamaño de la cabeza de un alfiler (1 a 3 mm), que son del color de la piel o ligeramente blanco-amarillentas.
Siringoma eruptivo. Predomina en pacientes jóvenes, habitualmente en el cuello, tronco, abdomen, muslos, nalgas y genitales. Se caracteriza por la aparición de múltiples erupciones de 2 a 3 mm, semiesféricas y aplanadas.
Siringoma diseminado o generalizado. Se caracteriza por la aparición de lesiones generalizadas predominantemente en tórax y región lumbar.
Siringoma aislado. Aparece una única lesión.
Siringoma lineal. Aparecen varias lesiones distribuidas linealmente.
Los siringomas a menudo se pueden confundir con otras afecciones de la piel, incluyendo el xantelasma , el tricoepitelioma múltiple, los quistes de millium, el granuloma anular, las verrugas planas, sarcoidosis e hiperplasia de las glándulas sebáceas, entre otras.
El especialista en dermatología deberá realizar las pruebas necesarias para conseguir un diagnóstico distintivo.
Siringomas en vulva y pene
En la vulva, se ven afectados los labios mayores de forma simétrica causando muchas veces un picor intenso. Se han descrito tres formas de presentación clínica: la más frecuente con múltiples pápulas simétricas, una variante milioide donde coexisten quistes de millium y siringomas y una tercera con aspecto de liquen simple crónico con eritema y erosiones.
En el pene suelen ser lesiones asintomáticas, múltiples o solitarias. Su localización más frecuente es el dorso y la cara lateral del cuerpo del pene. También pueden extenderse por la zona escrotal.
Tratamiento para eliminar los siringomas
Los siringomas son afecciones benignas que no requieren de tratamiento. Sin embargo, este se puede acometer para la eliminación de los siringomas por motivos meramente estéticos.
Entre los tratamientos disponibles para eliminar los siringomas se incluyen:
Electrocoagulación. Aplicación de una corriente eléctrica que coagula el tejido, quemando el siringoma.
Dermoabrasión. Se trata de lijar físicamente el tejido del siringoma y dejar que la piel vuelva a regenerarse.
Ablación con láser. El tejido es destruido mediante la aplicación de un láser.
Cirugía. En algunos casos, especialmente si los siringomas no están difuminados y se encuentran en zonas no comprometidas, también se puede recurrir a la eliminación mediante cirugía tradicional.
Se han utilizado tratamientos con isotretinoína, tretinoína, adapaleno o una solución acuosa de atropina tópica al 1%, aunque las recidivas o resurgimiento de los siringomas son frecuentes.
Ninguno de estos tratamientos se considera absolutamente satisfactorio y tampoco evitan las recidivas.
Artículo especializado
Betametasona: ¿Para qué se usa?
Los esteroides del grupo de los corticoides son un tipo de hormonas que se producen en las glándulas adrenales y tienen funciones en nuestro organismo especialmente a nivel del sistema cardiovascular, metabólico e inmunológico. La betametasona forma parte de los esteroides sintéticos.
¿Qué es la betametasona?
La betametasona es un fármaco glucocorticoide sintético de acción prolongada que actúa como inmunosupresor y antiinflamatorio.
Tras su toma, los efectos antiinflamatorios e inmunosupresores aparecen en unas 2 horas y se mantienen hasta 3-4 días.
Se puede administrar por vía oral, en forma de solución inyectable (intravenoso o intramuscular) o en pomadas de aplicación tópica. Sin embargo, en España no está comercializada la presentación oral de la betametasona.
¿En qué enfermedades se usa la betametasona?
La betametasona está indicada en múltiples patologías que cursan con inflamación:
Enfermedades reumatológicas. Incluyendo la artritis reumatoide, artritis gotosa, artritis psoriásica y en brotes de enfermedades autoinmunes.
Procesos inflamatorios articulares . Incluyendo sinovitis, bursitis, miositis, lumbalgias, ciatalgias, tortícolis o ganglión.
Enfermedades autoinmunes del colágeno. Se incluye el lupus eritematoso sistémico, esclerosis sistémica, dermatomiositis.
Enfermedades alérgicas. Incluyendo el asma, rinitis alérgica estacional, rinitis perenne, dermatitis de contacto, dermatitis alérgicas, reacciones de hipersensibilidad y picaduras de insectos.
Enfermedades de la piel . Incluyendo erupciones liqueniformes, liquen plano, psoriasis, lupus eritematoso sistémico, alopecia areata y queloide.
Afecciones del pie. Incluyendo la bursitis retrocalcánea, el espolón calcáneo, y las metatarsalgias.
En partos prematuros. Se puede utilizar para promover la maduración pulmonar del feto.
Enfermedades neoplásicas. Se puede administrar como tratamiento paliativo para disminuir la inflamación acompañando a la quimioterapia en casos de leucemia, linfomas, leucemia infantil linfoide y mieloide agudas.
Efectos adversos, contraindicaciones y precauciones de la betametasona
Los efectos secundarios de la betametasona se relacionan, especialmente, con su uso a largo plazo.
Efectos cardiovasculares . Aumento del riesgo de trombosis, tromboflebitis, arritmias cardiacas e hipertensión arterial.
Efectos dermatológicos. Pueden aparecer r eacciones de hipersensibilidad y decoloración o hiperpigmentación de la piel, irritación con quemazón y prurito, atrofia dermatológica, estrías, vitíligo, infecciones, hirsutismo, eritema facial, paniculitis, erupciones, exantemas y necrólisis epidérmica.
Efectos endocrinológicos . Aumento de glucosa en sangre, que puede dar lugar a la aparición de diabetes, síndrome de Cushing, aumento del peso corporal, amenorrea, trastornos del ciclo menstrual y alteraciones en el desarrollo con disminución del crecimiento en niños.
Disminución de electrolitos . Afectando a los niveles de potasio y calcio.
Trastornos Incluyendo náuseas, vómitos, dolor abdominal, pancreatitis, gastritis y esofagitis que pueden dar lugar a úlceras gástricas y hemorragias digestivas.
Disminución de la inmunidad. El uso crónico puede ocasionar, como consecuencia de una disminución de las defensas, la aparición de infecciones.
Efectos osteomusculares . Se puede propiciar la osteoporosis, fracturas óseas, miopatías, miastenia, daños en los tendones como tenosinovitis y ruptura tendinosas, osteonecrosis, en especial, en cabeza de fémur y húmero.
Afectación del sistema nervioso central. Puede producir o empeorar trastornos psicológicos como inestabilidad emocional, cambios de humor, euforia, agitación y tendencias psicóticas. Además, pueden causar cefalea, vértigo e insomnio.
Afectación infantil . En niños sometidos a tratamiento tópico se han notificado supresión del eje hipotálamo-hipófisis-suprarrenal, síndrome de Cushing, retraso del desarrollo e hipertensión intracraneal.
Contraindicaciones de la betametasona
La betametasona está contraindicada durante el embarazo, salvo en el caso de amenaza de parto prematuro entre las semanas 24 y 36 del embarazo para estimular la maduración pulmonar del feto.
La betametasona se excreta por la leche materna por lo que está contraindicada en mujeres lactantes.
Las formas tópicas de betametasona están contraindicadas en menores de un año. Tampoco debe emplearse en los ojos ni en heridas abiertas profundas.
Interacción de la betametasona con otros fármacos
Algunas de las interacciones farmacológicas que se pueden dar incluyen:
Su uso junto con las vacunas de virus atenuados de sarampión, rubéola, de bacterias como la salmonella, produce una disminución de la inmunidad de las vacunas, es decir una disminución de la generación de anticuerpos tras la vacunación.
No se debe usar junto con fármacos antiinflamatorios , ni aspirina, por el mayor riesgo de hemorragias gastrointestinales al usar ambos fármacos.
El uso junto con adenosina puede potenciar la toxicidad cardiaca de esta última.
El uso de betametasona junto con aldocumar puede dar lugar a alteración en el tiempo hemorrágico, causando un mayor riesgo de hemorragias.
El uso junto a antibióticos como la eritromicina aumenta el efecto de los glucocorticoides.
El uso con salbutamol puede dar lugar a un mayor número de arritmias cardiacas.
Los tópicos gastrointestinales (sales, óxidos e hidróxidos de magnesio, aluminio y calcio) pueden disminuir la absorción digestiva de los glucocorticoides.
Preguntas frecuentes
¿Cuánto tiempo se puede usar la betametasona?
La betametasona se debe administrar siempre a la menor dosis posible y el tiempo estrictamente necesario, siempre bajo la supervisión de un especialista.
En situaciones como el asma , se darán dosis de duración corta para controlar los posibles efectos secundarios de la betametasona.
¿Se puede usar la betametasona en niños?
La betametasona puede usarse en niños, pero en ellos se usa principalmente como parte del tratamiento de leucemias y linfomas.
El uso para otras enfermedades en niños es poco frecuente, ya que puede producir alteración del crecimiento. Hay siempre que valorar el riesgo-beneficio del fármaco cuando se inicia el tratamiento.
¿Es más peligrosa la betametasona en niños o en adultos?
La betametasona produce efectos secundarios tanto en niños como en adultos, pero en los primeros, además de efectos secundarios similares a los adultos, puede dar lugar a alteraciones en el crecimiento.

Artículo especializado
Gonalgia o dolor de rodilla: tratamientos
Las complejas características anatómicas de la rodilla determinan que se trate de una articulación frecuentemente afectada por traumatismos, así como por procesos inflamatorios y degenerativos. El dolor es un síntoma también frecuente en la rodilla.
¿Qué es la gonalgia?
La gonalgia es un término general que hace referencia a la aparición de dolor en la rodilla. Este puede estar causado por múltiples factores, incluyendo los de naturaleza traumática. Afecta a un 20-25% aproximadamente de la población adulta y su prevalencia ha aumentado un 65% en los últimos 20 años. No en vano, la rodilla es una de las estructuras del aparato locomotor más complejas, debido a la cantidad de estructuras que la componen. Juega una importancia primordial en la estabilidad y el desplazamiento, viéndose sometida a fuertes cargas y rápidos cambios de dirección.
La gonalgia puede aparecer junto a otros síntomas y signos:
Inflamación y rigidez
Enrojecimiento y aumento de la temperatura al tacto.
Sonidos de chasquidos, crujidos o sensación de bloqueo.
Incapacidad de realizar determinados movimientos con la articulación.
Las causas de la gonalgia se podrían dividir en dos grandes grupos:
Origen traumático . El traumatismo directo, la torsión excesiva de rodilla con pie fijo en suelo o la caída con golpe sobre las rodillas pueden producir desde una simple contusión, pasando por una lesión de ligamentos y meniscos, hasta una lesión ósea o procesos de recuperación tras una cirugía.
Origen no traumático . El dolor en la rodilla puede deberse a enfermedades como los procesos degenerativos, reumáticos o infecciosos. Entre ellos destacan la artrosis , artritis reumatoide (se produce destrucción de la articulación debido a mecanismos autoinmunes), gota o acumulación de cristales de ácido úrico y pseudogota por cristales de calcio, y artritis séptica por infección directa del espacio
Posibles causas de gonalgia
La gonalgia puede ir asociada a una gran variedad de lesiones de rodilla afectando a cualquiera de los tejidos que forman parte de ella, incluyendo ligamentos, tendones o bolsas sinoviales de la articulación. Es por ello que una parte fundamental del proceso será realizar un diagnóstico diferencial exhaustivo. Dentro de estas lesiones las más habituales en consulta suelen ser:
Artrosis . Su existencia suele estar relacionada con la edad, aunque existen muchos otros factores. Es más común en adultos de entre 70 y 74 años. Los síntomas principales son el dolor que puede empeorar con el inicio de la marcha y la limitación del rango articular de la rodilla. Se trata de una patología de evolución progresiva
Procesos inflamatorios articulares . El signo característico es la tumefacción (inflamación). El examen físico puede mostrar si el proceso inflamatorio es extra o intraarticular.
Bursitis . Se trata de lesiones localizadas bien delimitadas en el caso de la Pueden aparecer en la zona anterior de la rodilla (bursitis pre rotuliana) o en la zona lateral (bursitis anserina).
Tenidopatía rotuliana . Un exceso de cargas repetitivas sobre el aparto extensor de la rodilla puede llegar a producir la aparición de los síntomas, que generalmente suele ser dolor en la cara anterior de la
Síndrome patelofemoral . Se trata de la principal causa de dolor en la cara anterior de la rodilla entre adolescentes y adultos menores de 60 años. Su causa es multifactorial y será fundamental realizar un buen examen y exploración.
Alteración intraarticular y ligamentosa . Suele acompañarse de antecedentes traumáticos y rotura de los ligamentos o meniscos de la rodilla. Esto origina derrame articular y sensación de inestabilidad, bloqueo de rodilla en semiflexión o incapacidad
Tratamiento de la gonalgia
El tratamiento de la gonalgia variará en función de su causa.
Fisioterapia
Las diferentes técnicas y herramientas de fisioterapia, así como una correcta administración y dosis de diferentes tipos de ejercicio y contracciones musculares en función de la patología, irán encaminadas en la reducción del dolor y en la rehabilitación de la lesión si fuera posible.
Tratamiento farmacológico
Tratamientos específicos . Medicamentos indicados propiamente para enfermedades como la gota o artritis reumatoide.
AINES . El uso y utilidad de los antiinflamatorios no esteroideos se limita a las fases agudas.
Inyecciones articulares . Puede incluir la inyección en la rodilla de corticoides, ácido hialurónico o plasma rico en plaquetas para reducir la inflamación y el dolor. También pueden mejorar la movilidad y favorecer la cicatrización.
Cirugía
En algunos casos, la cirugía es el único método para reparar el problema articular. Puede realizarse a través de artroscopia, con pequeñas incisiones, para reparar cartílago y ligamento. También puede realizarse cirugía abierta en el caso de reemplazo de rodilla.
Otras terapias
Otro tipo de terapias o estrategias para minimizar la gonalgia incluyen el reposo, la ortesis o empleo de plantillas especiales en algunas patologías degenerativas, vendajes funcionales, estimulación eléctrica y la disminución de peso.
Si una gonalgia no es tratada a tiempo puede ocasionar lesiones osteocondrales residuales y recurrentes con alteraciones en los mecanismos de extensión y flexión, lo que se traduce en importantes alteraciones de la movilidad y limitación funcional.
Preguntas frecuentes
¿Qué diferencia hay entre gonalgia y gonartrosis?
La gonalgia es un término que hace referencia al dolor en la articulación de la rodilla de forma general, generalmente como síntoma, en cambio, la gonartrosis, es una enfermedad articular crónica degenerativa y progresiva de la rodilla, que seguramente curse con gonalgia.
¿Se puede realizar ejercicio padeciendo gonalgia?
El ejercicio ha demostrado ser beneficioso en la mayoría de las lesiones de rodilla. Es importante −siempre que esté indicado para su patología− realizar los ejercicios sin dolor. La monitorización de la dosis, la carga y el dolor durante el ejercicio serán fundamentales para realizar una buena administración del mismo. Es recomendable que consulte con su médico o fisioterapeuta los distintos ejercicios y frecuencia, dado que una mala gestión puede ser perjudicial.
¿Cuáles son las pruebas complementarias en caso de gonalgia?
La mayoría de las patologías de rodilla pueden ser diagnosticados con la realización de una historia clínica, un examen físico y los estudios radiográficos pertinentes.
Tras realizar el examen físico articular, el médico traumatólogo puede indicar alguna de las siguientes pruebas diagnósticas:
Rayos Permite descartar enfermedades degenerativas y fracturas.
Tomografía axial computerizada (TAC). Este método permite obtener imágenes radiográficas desde diferentes planos.
Ecografía. Produce imágenes en tiempo real de las estructuras del tejido blando del interior y alrededor de la rodilla.
Resonancia Magnética. Puede ser de utilidad para lesiones de ligamentos, cartílagos, músculos, tendones y tejido óseo.
Análisis del líquido articular. El examen del líquido sinovial puede ser útil en diversos procesos como la artritis infecciosa y las artritis
Artículo especializado
Embarazo anembrionario: síntomas y causas
Existen diversas causas que pueden determinar que un embarazo acabe en aborto en las primeras semanas de gestación. Una de ellas es el embarazo anembrionario.
¿Qué es el embarazo anembrionario?
El embarazo anembrionario es un embarazo en el que se forma un saco gestacional, pero sin un embrión en su interior. Este tipo de embarazo terminará en aborto, debido a que, aunque el huevo fertilizado se implanta en el útero, no acaba de desarrollarse el embrión.
Se trata de un problema relativamente frecuente. Cerca de un 15 % de los embarazos que son detectados clínicamente acaban perdiéndose de forma espontánea, y en un tercio de ellos se debe a embarazos anembrionarios.
De este modo, la principal consecuencia para la mujer del embarazo anembrionario es que se producirá el aborto por la inviabilidad de la gestación.
Causas del embarazo anembrionario
La causa más habitual de los embarazos anembrionarios son las anomalías cromosómicas o genéticas que tienen lugar en el momento de la fecundación.
Algunas de las alteraciones cromosómicas más frecuentes son las trisomías, monosomías o poliploidías.
No existen factores relacionados con las características de la mujer o el hombre que puedan predecir con certeza que se producirá este tipo de gestación fallida. Sin embargo, los abortos espontáneos suelen ser más frecuentes en función de la edad de la madre. A partir de los 35 años se incrementa el riesgo de problemas en el embarazo. De igual modo, si se han producido abortos con anterioridad, el riesgo de nuevos abortos también es mayor.
Otros factores que muestran una posible asociación con los abortos espontáneos y los trastornos del embarazo están relacionados con niveles insuficientes de vitaminas del grupo B como el ácido fólico y vitamina K en la madre.
Síntomas del embarazo anembrionario y cómo detectarlo
En muchos casos el embarazo anembrionario puede darse sin síntomas y pasar totalmente desapercibido. Incluso, el aborto espontáneo puede expulsarse junto con la regla.
En el caso de realizar una prueba de embarazo, esta daría positivo, ya que la hormona detectada en dicha prueba es segregada por células del saco gestacional, aunque no aparezca un embrión en su interior.
En cuanto a los síntomas para la mujer en el transcurso de un embarazo anembrionario, estos serían los síntomas propios de un embarazo normal, incluyendo:
Ausencia de la menstruación.
Aumento de la sensibilidad mamaria.
Náuseas.
Etc.
Estos síntomas cesarían en el momento en el que tuviera lugar el aborto.
Por otro lado, la realización de una ecografía en etapas tempranas de la gestación podría ayudar a detectar un embarazo anembrionario.
La ecografía transvaginal permite detectar el embrión en torno a las 6 semanas de gestación. El diagnóstico de embarazo anembrionario se podrá establecer ante el hallazgo de un saco gestacional mayor de dos centímetros y en cuyo interior no se visualice el embrión.
¿El embarazo anembrionario puede ser recurrente?
No suele ser normal que se repita un embarazo anembrionario, aunque es posible que se pueda dar en más de una ocasión. Haber sufrido una gestación anembrionaria no supone un mayor riesgo de sufrir otra.
De este modo, se podría intentar una nueva gestación a los 2 o 3 meses desde que se produjera un embarazo anembrionario, en función de cómo se haya producido el proceso abortivo y de las indicaciones del ginecólogo.
En el caso de presentar varias pérdidas gestacionales, sería recomendable realizar un estudio para identificar los motivos que pudieran estar detrás de los abortos recurrentes e intentar ponerles remedio.

Artículo especializado
Desgarro perineal: ¿Cuándo se produce?
El parto vaginal se asocia con la posibilidad de desgarros perineales, afectando al 80-85% de las mujeres durante el parto.
¿Qué es el desgarro perineal?
El desgarro perineal es una solución de continuidad, esto es, un corte transversal que afectará a distintos músculos según su grado (asociado a la profundidad del corte).
De este modo, debido a la maniobra en la que la cabeza del bebé debe atravesar la abertura vaginal durante el parto, se puede producir una lesión en la zona perineal, ya sea un desgarro o una episiotomía realizada expresamente por el médico.
Además, el parto vaginal también se relaciona con lesiones de la musculatura perineal, del esfínter anal y lesiones del músculo elevador del ano, con mayor incidencia en el parto instrumental (parto a través de fórceps o ventosa) que en el parto espontáneo.
Grados de desgarro perineal
Los desgarros o traumas perineales se pueden clasificar en función de la profundidad de la región anatómica a la que afecten en:
Desgarros de primer grado. Afectan a la piel y mucosa vaginal.
Desgarros de segundo grado. Afectan a la musculatura, excluyendo el esfínter anal.
Desgarros de tercer grado (clasificación de Sultan):
a) Menos del 50% del espesor del esfínter anal externo.
b) Lesión del 50% o más del espesor del esfínter anal externo.
c) Lesión que afecta al esfínter anal externo y al interno.
Desgarros de cuarto grado. Desgarro del esfínter anal y la mucosa rectal.
Consecuencias del desgarro perineal
Cuando se produce un desgarro perineal es importante una correcta identificación del mismo, ya que de lo contrario pueden quedar secuelas como dispareunia o dolor durante el acto sexual, dolor perineal crónico, incontinencia urinaria o incontinencia fecal.
En el caso de la lesión del esfínter anal durante el parto, su relevancia radica en su relación con la incontinencia anal y con diferentes síntomas defecatorios, incluyendo la urgencia defecatoria y el dolor.
La incontinencia anal se define como la pérdida involuntaria de gases o heces, que suele estar precedida de una sensación de urgencia y que puede afectar de forma muy importante a la calidad de vida de la mujer.
Prevención, tratamiento y alivio del desgarro perineal
Es importante tener en cuenta durante el parto las actuaciones necesarias para prevenir el desgarro perineal durante el parto. De este modo, se pueden establecer las siguientes recomendaciones:
Proteger el periné . Para ello, se debe aprovechar la elasticidad del suelo pélvico y lograr la expulsión de la cabeza del bebé en máxima flexión.
Aplicación de compresas calientes y masaje perineal. Con la intención de aumentar la flexibilidad y reducir la tensión de la musculatura perineal (se recomienda a partir de la 33 semana de embarazo).
Uso restrictivo de la episiotomía . Se aconseja usar la episiotomía mediolateral derecha en lugar de la central. Esta debe tener un ángulo de al menos 60° para que sea protectora (por cada 6° que se horizontalice la incisión de la episiotomía, disminuye un 50% el riesgo de desgarros con peores consecuencias).
Entre los factores de riesgo para la aparición de lesiones perineales más graves (tercer y cuarto grado) se encuentran:
No haber tenido hijos anteriormente.
Peso fetal superior a 4 kg.
Parto instrumentado con fórceps.
Distocia de hombros.
Episiotomía media.
Presentación occípito-posterior.
Etapa expulsiva del parto superior a una hora.
Posición materna en litotomía o en cuclillas.
Entre estos factores, algunos son modificables, sobre todo la instrumentación del parto, la realización de episiotomía y la posición materna. Su modificación permite disminuir la incidencia de las lesiones anales.
Por otro lado, la episiotomía sistemática no ha demostrado ser útil para la prevención de los desgarros perineales, por esto se aconseja realizarla de forma restrictiva.
En cuanto a la reparación de los desgarros perineales, se recomienda su sutura una vez que se ha producido el parto, salvo que se trate de lesiones del periné anterior que no sangren ni distorsionen la anatomía y de la piel del periné o el epitelio vaginal si los bordes están próximos y no hay sangrado.
Finalmente, en algunos casos es recomendable algún tratamiento farmacológico. Es el caso de una profilaxis antibiótica intravenosa antes de la reparación del desgarro del esfínter anal y su prolongación durante 5-7 días.
También se puede establecer un tratamiento con laxantes en caso de desgarro del esfínter anal o de la mucosa rectal.
Desgarro perineal y embarazo
En el caso de mujeres que han tenido un desgarro perineal, especialmente de los grados más graves, a la hora de plantearse otro embarazo la preocupación se centra en la posibilidad de que esta lesión pueda repetirse y que aparezcan o se agraven los síntomas.
De este modo, después de haber padecido un desgarro de tercer o cuarto grado durante el parto, si la mujer se vuelve a quedar embarazada, se debería tener en cuenta:
Si la mujer está asintomática y mantiene la continencia, se puede recomendar un parto vaginal. El riesgo de repetir otra lesión de tercer o cuarto grado es relativamente baja. En cualquier caso, la incontinencia anal no se modifica por la realización de una cesárea. En el caso de pesos fetales por encima de 4 kg el riesgo de lesión aumenta notablemente.
Si la mujer presenta incontinencia anal, es probable que el embarazo aumente la intensidad de la sintomatología. Sin embargo, no hay evidencia de que la vía de parto influya en la evolución posterior.
En los casos en que la mujer haya presentado una incontinencia anal posparto, con cirugía exitosa posterior, es recomendable el parto por cesárea .

Artículo especializado
El hierro en el embarazo y su importancia
La dieta de la embarazada debe contener un aporte energético adecuado para asegurar su propia salud y la del feto, ya que, desde el punto de vista nutricional, la dependencia del feto del organismo materno es total.
El hierro es uno de los nutrientes importantes durante el embarazo debido al elevado riesgo de anemia en esta etapa, dada la mayor demanda del mismo por parte de la gestante. En general, los niveles de hierro del feto dependen de los niveles de la madre, siendo un elemento esencial para el desarrollo de la mayoría de los órganos en el feto y también para el desarrollo normal del cerebro.
Anemia en el embarazo
La anemia ferropénica es muy frecuente en las mujeres embarazadas, sobre todo en el segundo y tercer trimestre de la gestación y después del parto. Algunas estimaciones señalan que el déficit de hierro durante el embarazo afecta a una de cada cinco embarazadas, pudiendo llegar a una de cada tres durante el tercer trimestre de embarazo.
Por otro lado, en España el déficit de hierro supone el 90% del total de anemias en las gestantes. De este modo, muchas gestantes necesitarán suplementarse con hierro durante esta etapa, si bien una alimentación adecuada puede prevenir la anemia por deficiencia de hierro en el transcurso de la gestación.
La Sociedad Española de Ginecología y Obstetricia (SEGO) recomienda la suplementación de hierro desde el segundo trimestre hasta pasado un mes del parto.
Se debe asegurar el aporte de 30 mg de hierro al día durante el embarazo en las gestaciones únicas y 60 mg/día en las gestaciones múltiples. Durante la lactancia el aporte debe ser de 15 mg/día.
Alimentos con hierro para el embarazo
Una dieta adecuada durante el embarazo debe incluir alimentos ricos en hierro, como carnes rojas magras, carne de ave, pescado y moluscos. Además, las opciones vegetales comprenden sobre todo las legumbres y verduras como espinacas y acelgas y los frutos secos.
El hierro que proviene de los productos de origen animal (hierro hemo), se absorbe mejor que el de origen vegetal. Para aumentar la absorción de hierro de las fuentes vegetales y de los suplementos es recomendable consumirlos junto con alimentos ricos en vitamina C (naranjas, tomates, fresas, kiwi, pimientos, etc.).
Para poder prevenir y tratar la anemia ferropénica, Savia pone a tu disposición los mejores especialistas en Endocrinología y Nutrición .
Síntomas de hierro bajo en el embarazo
Los síntomas de unos niveles bajos de hierro sin llegar a anemia son inespecíficos. La disminución de las reservas corporales de hierro puede dar lugar a la aparición de fatiga.
Por otro lado, cuando se desarrolla anemia por deficiencia de hierro (anemia ferropénica), los síntomas son más específicos y pueden aumentar con la severidad de la anemia.
En cualquier caso, los síntomas de la anemia, sobre todo en casos leves, pueden ser muy parecidos a los síntomas generales que se experimentan por el propio embarazo. De este modo, una mujer embarazada con niveles bajos de hierro puede experimentar:
Cansancio y debilidad.
Palidez de la piel y las mucosas.
Mareos.
Deterioro de la capacidad cognitiva.
Inestabilidad emocional y síntomas depresivos.
Mayor riesgo de infecciones.
En casos más graves, pueden aparecer palpitaciones, dificultad para respirar y dolor en el pecho.
Los análisis de hierro y hemoglobina, la principal proteína que lo contiene, serán habituales a lo largo del embarazo, junto con otros controles y análisis .
Síntomas de hierro alto en el embarazo
Tomar demasiado hierro durante el embarazo también puede ser contraproducente para la madre y el feto.
Algunos de los efectos relacionados con un exceso de hierro incluyen mayor riesgo de:
Preeclampsia.
Hipertensión arterial.
Diabetes mellitus.
Abortos espontáneos.
Consecuencias de la anemia en el embarazo
La anemia por deficiencia de hierro durante la gestación puede tener consecuencias negativas para la salud de las mujeres, impidiendo la realización normal de sus actividades cotidianas.
Las formas más graves de anemia pueden tener una influencia negativa en la gestación, ya que existe un mayor riesgo de aborto y de parto pretérmino, bajo peso al nacer (provocando, a su vez, un aumento de la mortalidad perinatal), así como un incremento del riesgo de infecciones puerperales.
Adicionalmente, la deficiencia de hierro en las madres, especialmente a principios del embarazo, está significativamente asociada con parto prematuro, peso bajo del recién nacido, peso bajo para la edad gestacional y una mayor mortalidad perinatal del neonato.
La deficiencia de hierro y la anemia durante la gestación inevitablemente se agravarán después de dar a luz (anemia posparto), debido a las pérdidas de sangre asociadas con el parto. Se estima que la prevalencia de anemia en el posparto es del 50% dentro de las 48 horas siguientes al parto. La mayoría de las formas serían prevenibles con una gestante que llegase al último trimestre de la gestación con sus reservas férricas en óptimas condiciones (hemoglobina mayor o igual a 11mg/dl).
Por otro lado, la deficiencia de hierro en la gestante afectará negativamente las interacciones entre la madre y el niño. Además, los niños nacidos de madres con deficiencia de hierro tienen un menor desarrollo cognitivo, motor, social, emocional y neurofisiológico de las funciones cerebrales.
En definitiva, después de analizar los resultados de los análisis correspondientes, el ginecólogo podrá recomendar la necesidad de suplementos de hierro y vitaminas, algunas de ellas implicadas en el metabolismo del hierro.

Artículo especializado
Sonda vesical: ¿Qué es y qué tipos existen?
La sonda vesical sirve para ayudar a drenar la vejiga y expulsar la orina que se almacena en ella. Se utiliza en diversas situaciones clínicas y en diferentes tipos de pacientes. Es una técnica invasiva, donde se introduce una sonda a través del meato urinario hasta la vejiga.
¿Qué es una sonda vesical?
La sonda vesical es un tubo elástico y fino que se introduce hasta la vejiga con el fin de establecer una vía de drenaje hasta el exterior con fines terapéuticos o diagnósticos. En función de las características del paciente y del fin terapéutico se pueden encontrar diferentes tipos de sondas.
Tipos de sondas vesicales
Existen diversos tipos de sondas destinadas a diferentes indicaciones clínicas y diferentes duraciones del sondaje.
Según el tiempo de permanencia, la sonda puede ser:
Sonda permanente . La sonda se deja introducida durante un periodo de tiempo variable que puede llegar a ser superior a los 30 días.
Sonda intermitente o evacuador . El catéter se retira en cuanto se ha vaciado la vejiga de orina.
Según su composición la sonda puede ser de:
Silicona . Presenta la mayor biocompatibilidad y mayor calibre funcional. Son las más finas y mejor toleradas. Es la más adecuada para el sondaje permanente.
PVC (cloruro de polivinilo) . Es un material más rígido. Se emplea en cateterismos intermitentes.
Látex . Pueden producir estrechamiento uretral, por lo que cada vez se utilizan menos. Su duración puede ser hasta 45 días.
Las sondas también pueden variar según el calibre. Para ello se utiliza la escala de Charriere . Una unidad o CH equivale a 0,33 mm. Los calibres disponibles se escalonan de dos en dos. En la elección del calibre de la sonda se tendrá en cuenta el diámetro de la uretra. Las medidas más habitualmente empleadas son los siguientes:
La sonda vesical femenina suele presentar un calibre de CH 14-16.
En la sonda vesical masculina el calibre suele ser de CH 16-18-20-22.
La longitud varía dependiendo del tamaño de la uretra (varón, mujer o niños) y del propósito del cateterismo y se expresa en centímetros o en pulgadas (una pulgada equivale a 25 mm). La longitud más habitual suele ser 41 cm.
Por otro lado, existen diversos tipos de sonda en función de la forma de la punta, que vendrá determinada por las circunstancias del paciente:
Dufour . Punta curva y con amplio orificio. Se usa para lavados vesicales con coágulos y orina con sangre.
Tiemann . Punta curva y redondeada. Para pacientes prostáticos, estenosis de uretra o dificultad de vaciado uretral.
Couvelaire . Punta biselada o en pico de flauta. Facilita el drenaje en pacientes con hematuria y/o para mantener un circuito de lavado.
Foley . Sondas rectas y con 2 o 3 luces. Flexibles con punta redondeada. Pueden ser de látex o silicona. Son las más utilizadas en general, para vaciado de vejiga y sondajes permanentes.
Mercier . Punta acodada. Para pacientes prostáticos y estenosis de uretra.
Sonda intermitente (Robinson, Nelaton). Existen distintos tamaños. Son catéteres rectos y semirrígidos. Se usa para vaciar la vejiga o la recogida de muestras.
Finalmente, el sistema de drenaje de la orina que se utiliza junto con la sonda puede ser:
Drenaje abierto. Precisa desconectar la sonda de la bolsa para realizar un vaciado o el cambio de la bolsa.
Drenaje cerrado. La sonda no se desconecta de la bolsa, ya que esta dispone de una llave o grifo. Es el más completo y seguro, ya que dispone de varios mecanismos que evitan la contaminación bacteriana.
¿Cuándo se utilizan las sondas vesicales?
Las indicaciones para la utilización de una sonda vesical pueden ser muy variadas, incluyendo:
Manejo de la retención aguda de orina.
Evaluación y control de la diuresis.
Tratamiento intraoperatorio y postoperatorio de intervenciones quirúrgicas prolongadas o en pacientes de riesgo previsible de retención de orina.
Para favorecer la cicatrización de heridas o escaras en pacientes con incontinencia.
Pacientes con inmovilización prolongada.
Para la mejora del confort en cuidados paliativos.
Tratamiento de algunos pacientes con vejiga neurógena.
Administración de terapias endovesicales.
Postoperatorio de algunas cirugías (uretra, próstata y vejiga).
Fístulas vesicales y rotura vesical extraperitoneal.
Hematuria de origen vesicoprostático en pacientes que requieran lavados vesicales continuos.
Además, el sondaje intermitente se debe emplear para:
Aliviar de forma inmediata la distensión vesical aguda.
Obtener una muestra de orina en pacientes con incontinencia.
Administración de fármacos (quimioterapia vesical, contraste radiológico, etc.).
Medir el volumen residual postmiccional.
Las contraindicaciones son las siguientes:
Prostatitis aguda.
Uretritis aguda y abscesos periuretrales.
Estenosis o rigidez uretral.
Sospecha de rotura uretral traumática.
Alergia conocida a los anestésicos locales o al látex.
Cuidados generales del paciente portador de sonda urinaria temporal o permanente
Lavarse las manos siempre antes y después de manipular la sonda y/o la bolsa colectora.
Realizar diariamente higiene y secado de genitales, evitando tirar de la sonda.
Cambiar o vaciar la bolsa antes de que esté completamente llena.
Se vigilará que el tubo y la bolsa colectora no sobrepasen el nivel de la vejiga. Así evitaremos el reflujo.
Evitar tirones que puedan provocar traumatismos o desconexiones accidentales del sistema.
Evitar que se formen acodaduras.
En caso de pérdida de orina, cambios en la orina (olor, color y cantidad), fiebre o dolor abdominal o lumbar se debe contactar con la enfermera o médico de familia del centro de salud.
Según el material de la sonda se recomienda una periodicidad concreta de cambio del catéter siendo las de silicona las que ofrecen la posibilidad de uso más prolongado, aunque siempre es aconsejable respetar las recomendaciones del fabricante, ya que cada marca tiene sus peculiaridades.

Artículo especializado
Mancha mongólica en el recién nacido
La mancha mongólica de nacimiento se muestra de color azulado en la parte inferior de la espalda del recién nacido. Se trata de la lesión pigmentada más frecuente en recién nacidos. Normalmente es de carácter benigno, aunque en ocasiones puede estar relacionada con algunos síndromes y trastornos metabólicos.
¿Qué es la mancha mongólica?
Las manchas mongólicas son la forma coloquial de denominar a la melanocitosis dérmica congénita . Se trata de manchas cutáneas de color azulado en la parte inferior de la espalda, presentes ya en el nacimiento, aunque también pueden aparecen durante las primeras semanas de vida.
La denominación de “manchas mongólicas” responde a que se trata de un tipo de manchas muy frecuentes en recién nacidos de piel negra o en asiáticos. En países como Mongolia su prevalencia es muy elevada, siendo mucho menos frecuente en niños caucásicos.
La mancha se muestra de un color gris azulado o violáceo porque el pigmento causante, la melanina, se encuentra localizada en estos casos en la dermis, la capa interna de la piel.
El tamaño puede ser muy variable, desde pocos milímetros, hasta más de 20 cm con bordes indefinidos y puede ser única o múltiple.
La localización más habitual es la glúteo-sacra, si bien las manchas también pueden aparecer en zonas como los hombros.
Cuando se presenta una localización atípica, existe una mayor probabilidad de que se produzca su persistencia en la edad adulta.
¿Cuándo se va la mancha mongólica?
Las manchas hiperpigmentadas de la melanocitosis dérmica congénita son algo más frecuentes en varones y en recién nacidos pretérmino. Las lesiones pueden alcanzar su mayor visibilidad al año de vida, momento en el que comienza su atenuación. Finalmente, desaparecen a lo largo de la infancia en casi todos los casos, normalmente entre el primer y el segundo año de vida. En casos excepcionales pueden permanecer hasta la edad adulta, especialmente cuando se trata de manchas múltiples que presentan una coloración más oscura, son mayores de 10 cm y su localización es diferente a la región lumbosacra.
¿Cómo diferenciar la mancha mongólica de otras alteraciones de la piel?
La mancha mongólica del recién nacido puede aparecer tras el nacimiento o durante las primeras semanas de vida. Su diagnóstico es clínico, por lo que no se precisan pruebas complementarias.
Los rasgos más característicos de las manchas mongólicas azules en recién nacidos incluyen:
Su color es gris azulado o violáceo.
Las manchas no presentan relieve.
Los bordes de las manchas no están definidos, sino que se trata de bordes irregulares.
El tamaño de las manchas suele ser mayor de 2 cm, llegando a alcanzar hasta los 20 cm.
Estas características deberán ser tenidas en cuenta por el pediatra para poder descartar los trastornos de la piel motivados por problemas metabólicos u otras alteraciones que cursas con distorsiones pigmentarias.
De este modo, en ocasiones las manchas mongólicas pueden ir asociadas a ciertos errores congénitos del metabolismo, especialmente a las enfermedades de depósito lisosomal como la gangliosidosis y la mucopolisacaridosis tipo 1 o enfermedad de Hurler. En estos casos, las manchas en recién nacidos abarcan una gran extensión, su pigmentación es intensa y se presentan de manera múltiple. Además, suelen ir asociadas con otros síntomas, incluyendo la hipotonía y las visceromegalias.
Otras patologías que pueden ir asociadas a las manchas mongólicas incluyen:
Hemangioma congénito.
Síndrome de Sturge-Weber.
Síndrome de Klippel-Trenaunay.
Cutis marmorata teleangiectásica congénita.
Mancha café con leche.
Facomatosis pigmentovascular.
En muchos casos, la presencia de manchas mongólicas se relaciona con un peor pronóstico de la enfermedad subyacente.
En cuanto al diagnóstico diferencial de las manchas mongólicas, deben descartarse otras afecciones que cursan con hiperpigmentación de la piel como:
Nevus azul. Se caracteriza por ser lesiones de tamaño pequeño y de bordes bien definidos.
Nevus de Ota. Las manchas aparecen dentro del ojo y en la piel que rodea el ojo.
Nevus de Ito. La hiperpigmentación se presenta en la zona del nervio acromioclavicular.
Finalmente, las localizaciones atípicas de la mancha mongólica pueden dar lugar a diagnósticos erróneos de maltrato o abuso infantil. Para distinguirlos, hay que tener en cuenta que, a diferencia de los hematomas, las manchas mongólicas no muestran cambios en su coloración, son indoloras y suelen tardar mucho más tiempo en desaparecer.
¿Cómo se trata la mancha mongólica?
En el momento del nacimiento, la extensión y coloración de la mancha mongólica y su posterior expansión pueden ser muy llamativas y alarmantes para los padres. Sin embargo, esta afectación de la piel suele ser de naturaleza benigna y tiende a desaparecer por sí sola con la edad.
Durante los primeros meses de vida las manchas pueden adquirir una coloración aún más acentuada, alcanzando la máxima coloración en torno a los dos años de vida. A partir de ese momento se suele ir atenuando. Finalmente, cerca de los 10 años de edad la mancha suele ser prácticamente imperceptible.
En conclusión, en condiciones normales la mancha mongólica no necesita ningún tratamiento específico. En el caso de que estén asociadas a otros trastornos y patologías, el tratamiento efectivo de estas puede determinar su desaparición.
Por otro lado, en el caso de manchas muy extensas o en localizaciones atípicas, es posible que se mantengan presentes durante más tiempo.
Artículo especializado
Sudamina: tratamiento y causas en adultos
La sudamina es una afección de la piel que suele ser frecuente en climas cálidos y húmedos y es más común en bebés dado que tienen una menor capacidad de regular su temperatura corporal. Sin embargo, también puede afectar a personas adultas, especialmente en individuos con una gran tendencia a la sudoración y en pacientes hospitalizados.
Qué es la sudamina
La sudamina o miliaria se produce cuando el sudor no puede salir al exterior ya que se queda atrapado debajo de la piel. Esta situación se produce debido a que se obstruyen los conductos que transportan el sudor de las glándulas sudoríparas a la superficie de la piel. Esta retención del sudor provoca una erupción con la aparición de ampollas pequeñas, además de inflamación, irritación y picor.
En el caso de los bebés, la erupción suele aparecer en el cuello, los hombros y el pecho. También puede producirse en las axilas, en el pliegue interno del codo y en las ingles. Es decir, suele localizarse en aquellas zonas de pliegues donde es más difícil que se evapore el sudor.
Sudamina en adultos
La sudamina o erupción por calor también puede afectar a los adultos, especialmente en condiciones de calor y humedad.
En los adultos tiende a producirse en zonas de contacto o pliegues de la piel (axilas, parte interna de los muslos, debajo de las mamas, etc.). También puede aparecer en zonas de la piel cubiertas o cuando se encuentran pegadas a una superficie durante un período prolongado de tiempo, como en personas que permanecen en cama por una patología de larga duración.
El tipo de sudamina puede variar en función de la capa de la piel en la que queda atrapado el sudor.
La forma más leve es la miliaria cristalina . Se produce cuando se obstruye el poro, que es la apertura del conducto sudoríparo en la superficie de la piel. Esta forma se caracteriza por la formación de pequeños granitos transparentes llenos de líquido que se rompen con facilidad. Es el tipo de afección que se conoce genéricamente como sudamina.
Por otro lado, la miliaria rubra se produce a un nivel más profundo de la piel. Con ella aparecen pequeñas ampollas con picor en el área afectada de la piel. Cuando las erupciones inflamadas de la sudamina se sobreinfectan y se llenan de pus tiene lugar la miliaria pustulosa .
Finalmente, la forma menos común de sudamina es la miliaria profunda . Esta afecta a la capa más interna de la piel, la dermis, provocando erupciones inflamadas que se acompañan de dolor y picor.
¿Qué causa la sudamina?
La sudamina o erupción por calor se produce cuando el sudor no llega a la superficie y queda atrapado debajo de la piel. Su manifestación más inmediata es la aparición de erupciones con pequeñas ampollas que pueden estar acompañadas de picor. En algunos casos pueden aparecer bultos profundos e inflamados.
La erupción generalmente desaparece una vez que la piel se enfría.
Existen algunos factores y situaciones que pueden aumentar el riesgo de padecer sudamina:
En recién nacidos. Los conductos sudoríparos inmaduros pueden predisponer a que se produzca sudamina.
Estar en climas cálidos y húmedos.
Realizar actividad física.
Permanecer en reposo o en cama durante periodos prolongados.
Estados febriles.
¿Cómo se cura la sudamina?
La sudamina suele ser una alteración benigna y transitoria que no precisa de un tratamiento específico. Puede tardar de 3 a 6 semanas en resolverse completamente, ya que la obstrucción de los conductos de las glándulas sudoríparas sólo desaparece cuando se renuevan las células que forman la estructura de los conductos.
Puede ser beneficioso mantener la piel fresca y seca, intentando evitar un entorno que incremente la sudoración. También debe evitarse el exceso de prendas de vestir que no permitan la transpiración de la piel.
De igual modo, es recomendable mantener una buena higiene de la piel, evitando obstruir aún más los conductos de las glándulas del sudor. Conviene evitar la utilización de polvos o cremas espesas.
En definitiva, es importante fomentar que el sudor pueda evaporarse y no quede atrapado en la piel. Los baños con agua templada pueden contribuir a mejorar el problema.
Tratamiento de la sudamina: cremas y remedios naturales
En los casos en los que exista un componente inflamatorio importante se pueden utilizar cremas o lociones con corticosteroides, teniendo siempre en cuenta que éstos deben utilizarse de forma puntual y durante el menor tiempo posible. En la mayoría de los casos la principal actuación debe consistir en mantener la piel fresca y seca y no suelen ser necesarios la cremas con corticosteroides.
Existen algunos remedios que pueden contribuir a minimizar la sudamina y la erupción que se produce con ella:
Utilizar paños mojados con agua fría sobre la piel o ducharse con agua tibia puede calmar la erupción que se produce con la sudamina.
Dejar que la piel se seque al aire.
Evita la utilización de cremas, cosméticos y otros productos que puedan contribuir al taponamiento de los poros de la piel.
Evitar utilizar cremas hidratantes con lanolina para evitar que los poros y los conductos sudoríparos se obstruyan.

Artículo especializado
Prueba de Romberg: ¿En qué consiste?
La prueba de Romberg es una prueba clínica que permite estudiar y detectar trastornos de la coordinación, del movimiento y del equilibrio.
¿Qué es la prueba de Romberg?
La prueba de Romberg (test de Romberg o maniobra de Romberg) es una prueba diagnóstica que sirve para identificar trastornos relacionados con la pérdida del equilibrio y de la coordinación motora o ataxia.
Es una exploración clínica que realiza el médico en personas con mareos, vértigos, caídas de repetición, inestabilidades y desequilibrios.
Sirve para estudiar el equilibrio estático .
¿En qué consiste la prueba de Romberg?
Consiste en la exploración de la estabilidad del paciente en posición de pie, con los pies juntos y los ojos cerrados, y la observación de la capacidad del mantenimiento del equilibro.
En primer lugar, se debe colocar al paciente en la posición adecuada: pies juntos, brazos alrededor del cuerpo y en bipedestación. El paciente debe permanecer quieto 30 segundos.
A continuación, se realiza la misma operación, pero con los ojos cerrados. Si la persona pierde el equilibrio, mueve los pies o extiende los brazos se considera prueba de Romberg positiva. Si el paciente permanece erguido sin desplazamiento se considera el resultado de la prueba negativo.
El médico valorará varios datos durante la prueba que pueden ayudar a orientar el diagnóstico:
La velocidad a la que se produce el desplazamiento.
El lado hacia el que se mueve. En un vértigo de origen periférico el paciente se caerá hacia el lado del vestíbulo afectado y en un vértigo central hacia atrás, un lado u otro, indistintamente.
Si se produce hacia adelante o hacia atrás.
El tiempo que tarda en perder el equilibrio desde que cierra los ojos (latencia). En un vértigo periférico existe un tiempo de latencia y en un vértigo central no.
Cuando no hay caída, se debe observar si el paciente se mantiene estable y sin moverse, o por el contrario se mueve para evitar la caída (ataxia estática).
Se trata de una prueba rápida y sencilla que debe ser realizada correctamente por un médico en un espacio amplio, sin obstáculos. Además, la persona no debe estar apoyada en ningún sitio. El médico observará al paciente en todo momento para interpretar la prueba y vigilar cualquier caída.
Otras versiones de la prueba de Romberg incluyen el “Romberg sensibilizado”. Se realiza igual con el paciente en bipedestación, pero con un pie delante del otro (tándem) y con los brazos cruzados. Si la persona pierde el equilibro se considera Romberg sensibilizado positivo.
También existe la prueba de “Romberg modificada”. Se realiza con el paciente de pie, con los brazos estirados al frente, con el hombro en un ángulo de 90◦ y las palmas de las manos hacia arriba; se le indica al individuo cerrar los ojos y que se apoye en un pie. Se mide el tiempo en segundos que se mantiene en posición monopodal. La prueba se repite con el miembro inferior contralateral.
¿Qué ocurre si se da positivo en la prueba de Romberg?
La prueba de Romberg mide la propiocepción de una persona, o la capacidad que tiene el cerebro de saber la posición exacta de las diferentes partes del cuerpo en cada momento y detectar el movimiento y la posición articular.
Para mantener el equilibrio se combinan:
La visión.
El sistema vestibular es esencial en el control del equilibrio. Da información al cerebro de los movimientos y posición del cuerpo y la cabeza. Incluye el aparato vestibular del oído interno, el VIII nervio craneal y los núcleos vestibulares del tronco encefálico y el cerebelo.
El control neuromuscular.
Existes diferentes enfermedades que pueden alterar la coordinación:
Vértigo posicional paroxístico benigno, neuronitis vestibular, enfermedad de Ménière y laberintitis.
Alteraciones tóxico-metabólicas, déficit de Vitamina B12, neuropatía periférica diabética.
Enfermedades del sistema nervioso central: tumores, accidentes cerebrovasculares.
Infecciones.
Ciertas enfermedades hereditarias como la ataxia de Friedreich.
Fármacos.
Registro craneocorpográfico
Se trata de un método sencillo en el que el paciente lleva colocado un casco con unas luces en la cabeza y en los hombros. Se realiza en un cuarto oscuro, un espejo convexo y una cámara instantánea enfocada hacia el espejo. Se utiliza para registrar movimientos durante la realización de diferentes test, entre ellos el de Romberg.
Permite el registro foto óptico sobre una película de los movimientos de la cabeza y del cuerpo. La huella fotográfica ofrecerá una imagen de la amplitud de las oscilaciones del centro de gravedad.
La prueba se considera normal si la representación foto óptica se circunscribe a un punto (no hay oscilaciones).
En el estudio de una persona con mareo, alternaciones del equilibro o problemas de coordinación es necesario realizar un examen neurológico completo. Dentro de este se realizará la prueba de Romberg. Si el medico considera la maniobra de Romberg positiva, con el fin de encontrar la causa se realizarán otros exámenes y estudios en disciplinas médicas como la Otorrinolaringología y la Neurología .

Artículo especializado
Cómo curar una herida infectada
Cuando se produce una herida es importante proporcionar los medios que permitan su curación sin cicatrices ni secuelas. Cuando la herida está infectada es necesario acudir al médico.
¿Cómo saber si una herida está infectada?
La infección de una herida se produce cuando bacterias u otros microbios penetran en la ruptura de la piel y comienzan a multiplicarse. Nuestro sistema inmune responde a esta invasión para frenar esta infección .
Existe una mayor probabilidad de que la herida se infecte en determinados supuestos:
Heridas extensas y profundas.
Heridas por mordeduras.
Heridas sucias o contaminadas
Heridas de larga evolución.
Heridas con cuerpo extraño.
Cuando la persona que se ha hecho la herida padece diabetes, problemas circulatorios, problemas de cicatrización, un sistema inmunitario debilitado, desnutrición y abuso de alcohol y tabaco.
Por otro lado, existe una serie de síntomas y signos que se relacionan con la aparición de una infección y requieren la atención precoz por parte de los profesionales sanitarios, entre ellos:
Enrojecimiento.
Calor.
Hinchazón de la zona de la herida.
Secreciones purulentas de color amarillo-verdosas.
Mal olor, olor fétido.
Retraso en la cicatrización de la herida.
Aumento del tamaño de la herida.
Dolor nuevo o aumentado de la herida.
Aparición de fiebre o mal estado general.
En algunas ocasiones es necesario realizar cultivos microbiológicos de la herida para analizar si existe infección y determinar el microorganismo responsable. También nos puede indicar el antibiótico más adecuado para tratar cada infección concreta.
¿Qué hacer ante una herida?
Cuando se produce un corte poco profundo que sangra poco se recomienda:
Limpiar y desinfectar las manos que van a realizar la cura.
Lavar la herida con agua y jabón para retirar toda la suciedad posible.
Limpiar y secar la herida con gasas o paños los más asépticos que sea posible.
Aplicar un antiséptico (clorhexidina) y dejarlo secar.
Cubrir la totalidad de la herida con apósitos humedecidos o gasas y sujetarlos con vendas o esparadrapo. No tapar nunca la herida con papel higiénico ni material que pueda quedar adherido.
Revisar la herida de forma periódica para examinar la cicatrización y la posible infección o sangrado.
No utilizar algodón ni alcohol.
En caso de heridas muy sangrantes se debe hacer presión sobre la zona que sangra con gasas o paños limpios y solicitar ayuda sanitaria urgente.
¿Se puede aplicar alcohol en las heridas?
En primer lugar, se debe lavar bien la herida con agua y jabón para retirar toda la suciedad posible y, posteriormente, secar la herida con gasas o paños limpios.
En cuanto al alcohol, este es útil para desinfectar la piel sana (como antes de poner una vacuna o inyección), pero no debe usarse en las heridas abiertas, por su poder citotóxico para el nuevo tejido en formación.
Tratamiento de una herida infectada
Una herida que presenta una mala evolución, con una cicatrización deficiente o existen sospechas de infección, se hace necesario acudir al médico para que lleve a cabo una exploración y una valoración.
Si se diagnostica que la herida está infectada, el tratamiento puede incluir:
Antibióticos tópicos (mupiricina o ácido fusídico en crema).
Antibióticos orales (amoxicilina, amoxicilina-ácido clavulánico o clindamicina).
Antibióticos intravenosos.
Cirugía o curas locales específicas en función de las características de la herida y de la infección.
Si existe dolor o inflamación el médico puede recetar analgésicos o antiinflamatorios.
Algunas heridas requieren profilaxis antitetánica, por lo que en el centro sanitario se revisará el estado vacunal del paciente y se valorará la necesidad de recordatorio de vacuna antitetánica en función del riesgo.
Los antibióticos siempre deben ser prescritos por un médico. Se debe utilizar el más adecuado para cada infección y es muy importante cumplir las pautas de administración diarias y la duración del tratamiento. De lo contrario, el uso incorrecto de los antibióticos puede generar resistencias.
Cuidados tras la cicatrización
En el proceso de curación de las heridas intervienen varios procesos celulares y moleculares. La respuesta inmediata a la lesión es la vasoconstricción para minimizar el sangrado, que es causada por sustancias activadas en la sangre como prostaglandinas y tromboxanos. De igual modo, las plaquetas se adhieren al colágeno expuesto en la herida, mientras se activa a la cascada de coagulación. A partir de aquí se inicia el proceso de regeneración tisular y cicatrización.
Tras el cierre de una herida es importante cuidar la cicatriz para evitar complicaciones como la aparición de queloides (un crecimiento excesivo del tejido de una cicatriz) o que se produzca una hiperpigmentación de la región afectada por la cicatriz.
Algunas recomendaciones incluyen:
Evitar la exposición solar. Si la cicatriz va a estar expuesta al sol es necesario utilizar crema solar con factor de protección 50.
Mantener la zona de la herida hidratada.
Existen parches y lociones reductores de cicatrices e incluso en casos concretos puede ser necesario tratarla con láser.
Es importante observar la cicatriz y vigilar si la zona cambia de color, se inflama o aumenta de tamaño y comienza a ser dolorosa. En caso de que esto suceda será necesario consultar con el médico.
Si sospechas que una herida puede estar infectada acude a tu médico para que la valore y establezca el tratamiento antibiótico adecuado, si es preciso.
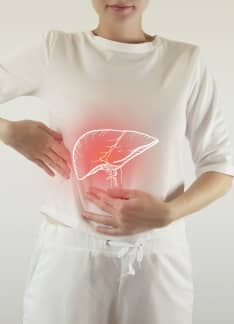
Artículo especializado
Síndrome de Gilbert: síntomas y diagnóstico
El síndrome de Gilbert es una enfermedad genética que afecta al hígado. Debido a que en ocasiones no provoca ningún síntoma, el número de las personas afectadas podría ser elevado.
¿Qué es el síndrome de Gilbert?
El síndrome de Gilbert es una alteración hepática que se da en familias, es decir, existe una condición hereditaria que se trasmite de manera autosómica recesiva y se asocia a un elevado nivel de bilirrubina en sangre (hiperbilirrubinemia no conjugada) causada por un déficit parcial de la enzima glucuroniltransferasa.
Este trastorno hepático es de causa y pronóstico benigno. Produce como síntoma principal una coloración amarillenta de la piel o “ictericia” que se desencadena a partir de un estímulo convencional (esfuerzo físico, estrés, una infección, etc.), que no conlleva ningún daño hepático asociado ni un mayor riesgo de padecer otras enfermedades.
En la mayoría de las ocasiones, el síndrome de Gilbert pasa desapercibido y es detectado de forma casual al realizar una analítica por otros motivos. Puede estar presente 10% de la población de Europa Occidental, principalmente del sexo masculino. Se diagnostica con mayor frecuencia durante la adolescencia y en adultos jóvenes entre los 20 y 30 años de edad, cuando se realiza una analítica de sangre rutinaria y aparece un nivel de bilirrubina alto, cuyos valores oscilan entre 20 y 80 mmol/dl.
El síndrome de Gilbert se produce por la disminución de la capacidad de excretar bilirrubina por el hepatocito (célula hepática). La bilirrubina surge de la degradación de la hemoglobina de los glóbulos rojos. Estos son producidos en la médula ósea y por lo general desempeñan su tarea de levar oxígeno a los tejidos durante cerca de 120 días.
Al destruirse los glóbulos rojos liberan la hemoglobina que es metabolizada. El grupo hemo se transforma en biliverdina y esta, a su vez, en bilirrubina «no conjugada» o indirecta. Al pasar por el hígado, esta bilirrubina reacciona con el ácido glucurónico gracias a la enzima uridindifosfoglucuronato glucuronosiltransferasa (UGT), transformándose en bilirrubina «conjugada» o directa. Esto es lo que permite que se disuelva en el agua y pueda ser excretada en la bilis.
La producción de la enzima UGT se regula por un promotor que es el que puede presentar la mutación y determina la producción de solo el 30 % de la cantidad normal.
Los adultos producen en el hígado entre 400 y 800 ml de bilis cada día. Además de bilirrubina, contiene colesterol y ácidos biliares, electrolitos (potasio, sodio y otras sales), metales como el cobre y agua. Los ácidos biliares en la bilis son importantes para la digestión y la absorción de grasas y vitaminas.
Por otro lado, la bilis también es importante para la eliminación de bilirrubina y otros materiales de desecho, que viajan en el fluido a través de la vesícula biliar y en el intestino delgado donde son convertidos en otras sustancias y, finalmente, expulsadas por medio de la orina y las heces.
Bilirrubina no conjugada elevada
En las personas con síndrome de Gilbert no toda la bilirrubina puede transformarse a la forma conjugada (soluble), lo que dificulta su excreción con la bilis y determina los niveles aumentados de bilirrubina no conjugada en sangre que desencadena la ictericia.
¿Cuáles son los síntomas y factores desencadenantes?
Mientras que la ictericia es el único síntoma clínico reconocido del síndrome de Gilbert (caracterizado por la aparición de una coloración amarillenta de la piel, la esclerótica y las conjuntivas de los ojos), algunas personas con la enfermedad han informado de la presencia de otros síntomas, incluyendo:
Cansancio y la fatiga.
Náuseas.
Poco apetito.
Dolor de estómago.
Síntomas relacionados con el síndrome intestino irritable (molestias gastrointestinales, estreñimiento, diarrea y distensión abdominal).
Problemas de concentración.
Orina de color oscuro.
Malestar general.
Se desconoce si estos síntomas pueden ser causados por el propio síndrome, ya que aún no se ha encontrado ningún vínculo entre ellos y el nivel de bilirrubina en la sangre.
Ciertos factores pueden ser desencadenantes de la ictericia en el síndrome de Gilbert. Entre ellos destacan:
Un esfuerzo físico excesivo.
Situaciones de estrés.
Ayuno prolongado o dietas hipocalóricas.
Infecciones.
Menstruación.
Deshidratación
Insomnio.
Cirugía.
El tratamiento con algunos medicamentos (quimioterápicos, anticonceptivos orales, etc.).
¿Cómo se diagnostica el síndrome de Gilbert?
El síndrome de Gilbert debe sospecharse siempre ante un paciente que presente:
Hiperbilirrubinemia ligera no conjugada.
Sin otros síntomas generales.
Sin manifestación de hemólisis.
Con una función hepática normal.
Tenga antecedentes familiares del síndrome.
Para el diagnóstico es suficiente en la mayoría de los casos la sospecha clínica y una analítica de sangre que muestre la elevación de la bilirrubina total a expensa de la no conjugada (casi siempre por debajo de 80 µ mol/L o 4,68 mg/d), con el resto de las pruebas hepáticas normales.
Dado que el síndrome de Gilbert no provoca más complicaciones, no precisa tratamiento.
El síndrome de Gilbert y alcohol
Al no determinar ningún problema hepático añadido, el síndrome de Gilbert no conlleva ninguna recomendación específica sobre el consumo de alcohol, más allá de las recomendaciones de moderación aplicables a la población general.
¿Qué aumenta los niveles de bilirrubina con el síndrome de Gilbert?
Se puede reducir la posibilidad de ictericia siempre que sea posible, tratar de evitar lo siguiente:
El ayuno o dietas muy bajas en calorías.
No beber suficientes líquidos.
Estrés y ansiedad.
Las infecciones.
El ejercicio vigoroso.
No dormir lo suficiente.
Artículo especializado
Índice de saturación de transferrina: ¿Qué es?
El índice de saturación de transferrina está incluido en una prueba analítica normal. Indica la capacidad de las proteínas sanguíneas de unirse al hierro para transportarlo.
¿Qué es el índice de saturación de transferrina?
El índice de saturación de transferrina muestra el porcentaje de transferrina que aparece en la sangre unida al hierro.
La transferrina es una proteína que se produce en el hígado, cuya función es transportar el hierro hacia los diferentes tejidos. Una vez sintetizada, se dirige a la médula ósea para la formación de glóbulos rojos y al bazo, el hígado y los músculos para su almacenamiento en forma de ferritina. Los valores de esta proteína son útiles para conocer el estado nutricional o la función hepática de una persona.
En función de diversos factores individuales como la actividad hepática y la alimentación, principalmente, la cantidad de transferrina puede variar.
El parámetro de saturación de la transferrina tiene en cuenta el hierro presente en el suero de la sangre, junto con:
La capacidad total de fijación de hierro (TIBC, por sus siglas en inglés)
La capacidad latente de fijación de hierro (UIBC, por sus siglas en inglés).
De este modo, el análisis de saturación de la transferrina en sangre se suele llevar a cabo junto con los valores de hierro en sangre y transferrina (o TIBC). También puede ser de utilidad el valor de ferritina, la proteína de almacenamiento del hierro.
Los valores normales de saturación de la transferrina pueden verse modificados en función de diversos factores individuales como:
La edad.
El sexo.
El tipo de alimentación.
Factores genéticos y poblacionales.
Los valores de saturación de transferrina deben valorarse junto con el historial médico de cada paciente y el resto exploraciones complementarias de las que se disponga.
Valores normales del Índice de Saturación de Transferrina
Los valores de saturación de transferrina que estarían dentro de lo normal serían:
Hombres: 20-50 %.
Mujeres: 15-50 %.
Niños: 17-44 %.
Recién nacidos: 56-74 %.
Síntomas de transferrina baja
El índice de saturación de transferrina mide el porcentaje de hierro transportado por la transferrina del total disponible.
Los valores bajos de saturación transferrina en sangre suelen indicar niveles de hierro bajos en el organismo. Esto sucede en algunos tipos de anemia, asociados a niveles bajos de hemoglobina.
De este modo, el tipo de anemia que se acompaña de niveles reducidos de saturación de la transferrina es la anemia ferropénica (por déficit de hierro).
En una anemia ferropénica leve los índices de saturación de transferrina están ligeramente disminuidos (10-20 % en hombres y 10-15 % en mujeres). En este caso, el resto de los parámetros mostrarían lo siguiente:
Los niveles de hierro en sangre suelen estar bajos (por debajo de 60 µg/dL).
La ferritina está baja.
El TIBC está alto (superior a 400 µg/dL).
En una anemia producida por una enfermedad crónica los valores podrían ser:
Niveles de hierro bajos.
La ferritina en valores normales o altos.
TIBC en valores normales o reducidos.
En una anemia ferropénica severa el índice de saturación de transferrina sería muy bajo (inferior al 10 % en adultos) con el resto de los parámetros del siguiente modo:
Niveles bajos de hierro en sangre (por debajo de 40 µg/dL).
Ferritina baja (inferior a 10 ng/ml).
Niveles altos de TIBC.
Además de la anemia ferropénica, otras situaciones que pueden producir una disminución de los valores de saturación de la transferrina en sangre incluyen:
Embarazo
Enfermedades hereditarias como la talasemia.
Enfermedades crónicas con pérdida de proteínas (infecciones, grandes quemaduras, etc.).
Patologías hepáticas y renales.
Algunos tipos de cáncer.
Alcoholismo.
Desnutrición.
De este modo, los síntomas que suelen acompañar a unos niveles de saturación de transferrina bajos incluyen:
Cansancio.
Debilidad.
Mareos.
Dolor de cabeza, etc.
Síntomas de transferrina alta
Unos valores elevados del índice de saturación de transferrina pueden señalar la presencia de hemocromatosis , una enfermedad hereditaria que provoca una acumulación de hierro en el organismo.
De este modo, una saturación de la transferrina por encima del 50 % puede ser un signo de la presencia de hemocromatosis.
Otras situaciones que pueden provocar un aumento de los valores saturación de la transferrina en sangre incluyen:
Intoxicación por hierro.
Transfusión múltiple de sangre.
Anemia sideroblástica.
Anemia hemolítica.
Anemia megaloblástica.
Cirrosis hepática.
Los síntomas que pueden aparecer con unos niveles de saturación de la transferrina elevados, debidos principalmente a la acumulación de hierro en el organismo, pueden incluir:
Fatiga generalizada.
Dolor articular.
Dolor abdominal.
Pérdida de peso.
Disminución del apetito sexual.

Artículo especializado
Ácido Úrico: ¿Qué significa su alta o baja presencia en sangre?
El ácido úrico o urato, dependiendo de sus niveles en la sangre, puede producir determinadas patologías como la gota.
¿Qué es el urato?
El urato o ácido úrico es el producto de la degradación de las purinas, compuestos que forman parte del ADN de las células. Una concentración elevada de ácido úrico en la sangre puede provocar el depósito de cristales de urato sódico en las articulaciones, provocando su inflamación y dando lugar a la aparición de gota (podagra). En esta patología las articulaciones sufren inflamación y dolor.
El ácido úrico es sinónimo de urato. Debido al pH fisiológico, la casi totalidad de moléculas del organismo están en forma de urato (la sal del ácido úrico). Por lo tanto, urato y ácido úrico es lo mismo.
Los niveles séricos de urato vienen determinados por el balance entre sus tasas de producción y eliminación. Aproximadamente, el 70 % del urato producido diariamente es excretado por el riñón a través de la orina. El resto es eliminado en el tracto biliar, vía intestinal. De este modo, una disminución de la eliminación de urato o un aumento en su producción pueden causar hiperuricemia.
Valores normales de ácido úrico
Los valores normales de urato en sangre son:
Hombres: de 3,4 a 7 mg/dl.
Mujeres: de 2,4 a 6 mg/dl.
Urato alto en sangre
El motivo de la gran mayoría de los casos de hiperuricemia es un defecto en su eliminación renal.
La existencia de un nivel elevado de urato en sangre es un escenario propicio para que se produzca la gota, con un aumento del riesgo a medida que lo hace la uricemia.
La hiperuricemia y la gota han ido aumentando sus tasas de prevalencia de forma significativa en las últimas décadas en las sociedades desarrolladas, debido principalmente a los cambios en la dieta, a través de alimentos especialmente ricos en proteínas (con elevado contenido en purinas).
Además de los factores dietéticos, otros factores pueden influir en el aumento del urato en sangre, incluyendo:
La edad.
Algunos medicamentos (diuréticos y bajas dosis de aspirina).
La enfermedad renal crónica.
Síndrome metabólico y obesidad.
Alteraciones genéticas en la síntesis y excreción renal del ácido úrico.
Además, la hiperuricemia y la gota aparecen relacionadas con otras patologías, incluyendo:
Litiasis renal o piedras en el riñón.
Hipertensión arterial.
Diabetes mellitus.
Hiperlipidemia y aumento del riesgo cardiovascular.
Alimentos y ácido úrico en sangre
Existe diversidad de alimentos que se relacionan con los niveles de ácido úrico en sangre. En unos casos para aumentarlos y en otros para contribuir a su reducción.
Entre los alimentos que aumentan los niveles de urato en sangre se encuentran:
Alimentos ricos en purinas . Destacan las carnes (incluyendo vísceras, procesados y extractos), los pescados y mariscos y algunos vegetales (legumbres, espinacas, espárragos y setas).
Alcohol . El consumo de bebidas alcohólicas (especialmente la cerveza), tanto en hombres como en mujeres, se asocia con niveles más altos de uricemia.
Entre los alimentos que pueden contribuir a reducir los niveles de ácido úrico en sangre se encuentran:
Los lácteos (especialmente desnatados).
Las verduras.
Alimentos ricos en vitamina C (fresas, cítricos, pimientos, etc.).
El café.
La fibra dietética (alimentos vegetales en general).
Las cerezas.
Finalmente, son bajos en purinas, por lo que tendrían un efecto neutro:
Los productos lácteos (leche, queso, yogur, helados).
Los huevos
Los cereales y sus derivados (pan, pasta, etc.).
Las verduras (salvo espinacas y espárragos).
Las frutas.
Los frutos secos.
Azúcares y dulces.
Urato bajo en sangre
Los niveles reducidos de urato en sangre o hipouricemia tiene lugar cuando los niveles en sangre de ácido úrico están por debajo de 2 mg/dl. Suele acontecer cuando se produce una eliminación excesiva de ácido úrico en la orina.
La hipouricemia no provoca sintomatología y, por lo tanto, no requiere tratamiento.
Una patología que puede causar niveles disminuidos de urato en sangre es la hipouricemia renal hereditaria, un trastorno genético que afecta al riñón caracterizado por la pérdida de urato en la orina. Puede determinar una mayor predisposición a la litiasis y puede causar insuficiencia renal aguda inducida por el ejercicio.
Otras patologías renales que pueden cursar con hipouricemia incluyen:
El síndrome de Toni-Debré-Fanconi.
El síndrome de Lowe.
La intoxicación por metales pesados.
La nutrición parenteral.
La enfermedad de Hodgkin y otras neoplasias.
La enfermedad de Wilson y otras causas de cirrosis.
La diabetes mellitus.
El síndrome de secreción inadecuada de ADH.
Hiperparatiroidismo.
La hiponatremia inducida por tiazidas (tipo de diurético).
La hiperbilirrubinemia.
Finalmente, algunos medicamentos que pueden aumentar la excreción urinaria de ácido úrico incluyen los estrógenos, el losartán (hipotensor), los contrastes radiológicos intravenosos, el dicumarol, los salicilatos a altas dosis y la trimetoprima-sulfametoxazol, entre otros.
Los niveles de ácido úrico o urato en sangre se pueden conocer gracias a una analítica clínica básica .
Uratos amorfos en orina
El urato amorfo o la presencia de cristales de uratos amorfos en orina es un parámetro del análisis de orina que puede significar una predisposición a la formación de cálculos renales.
Los cristales de uratos amorfos no causan síntomas, por lo que se suelen identificar cuando se realiza de forma rutinaria un análisis de orina .
Los uratos amorfos son visualizados mediante un microscopio y se relacionan con un pH ácido de la orina. Además, otras situaciones que pueden provocar la formación de urato amorfo y otros cristales en la orina son:
Un alto consumo de proteínas en la dieta.
Deshidratación o bajo consumo de agua.
Hiperuricemia y gota.
Algunas patologías renales.
Litiasis renal.
Ciertas patologías hepáticas.
Dieta rica en calcio o vitamina C.
No debe establecerse un tratamiento para la presencia de urato amorfo en orina, sino para la causa que la provoca.

Artículo especializado
CHCM: ¿Qué es y qué indica en tu analítica?
El CHCM es un índice analítico que sirve para valorar la cantidad de hemoglobina que está presente en la sangre.
¿Qué es el CHCM?
CHCM corresponde a las siglas de “Concentración de Hemoglobina Corpuscular Media”. Es un parámetro que representa la concentración media de hemoglobina que contiene cada glóbulo rojo. La hemoglobina es la proteína de los glóbulos rojos que lleva oxígeno de los pulmones al resto del cuerpo.
Para el cálculo del CHCM se toma el valor del contenido de hemoglobina en una muestra y se divide por el hematocrito, que es el volumen que ocupan los glóbulos rojos presentes en esa muestra sanguínea.
Los analizadores automáticos que se utilizan hoy en día para los análisis de sangre realizan los recuentos celulares y de hemoglobina utilizando diferentes métodos como la impedancia o la difracción de la luz, entre otros, y sus sistemas de cálculo integrado permiten obtener los índices eritrocitarios, incluyendo el CHCM de forma automática.
El estudio de los índices eritrocitarios, como el volumen corpuscular medio (VCM), el CHCM y el recuento reticulocitario (que mide la velocidad de renovación de los glóbulos rojos) en el contexto de unas de las patologías hematológicas más frecuentes como es la anemia, nos permite clasificarla en:
Normocítica-normocrómica.
Microcítica-hipocrómica.
Macrocítica.
Regenerativa.
Arregenerativa.
Estos parámetros constituyen la base del diagnóstico de la anemia que está asociada a síntomas como:
Debilidad o cansancio.
Fatiga.
Palidez.
Taquicardias.
Manos o pies fríos.
Problemas respiratorios.
Por otro lado, los valores normales de CHCM se sitúan en:
Adultos: 32-36 g/dl.
Niños: 31-37 g/dl.
¿Qué indican unos valores de CHCM bajos?
Dentro de los resultados de un análisis de sangre , un valor de CHCM bajo se corresponde con una anemia microcítica. Este trastorno hematológico se caracteriza por la presencia de eritrocitos de tamaño inferior a lo normal (con un VCM bajo) y asociado a una hemoglobina corpuscular media (HCM) y una CHCM bajas (hipocromía).
La causa más habitual de la anemia microcítica hipocrómica es la ferropenia o niveles bajos de hierro en el organismo. Esto es así en más de un 95% de las ocasiones. En cualquier caso, la confirmación deberá respaldarse con la historia clínica del paciente y el estudio de parámetros relacionados con el metabolismo del hierro.
Sin embargo, el CHCM se trata de un parámetro poco específico para valorar las anemias ferropénicas, ya que sólo en una parte minoritaria se aprecian valores disminuidos de CHCM. Para el diagnóstico de la anemia ferropénica existen otros parámetros más adecuados, incluyendo los valores de hemoglobina, el VCM, el RDW, el hierro total sérico, o los niveles de las proteínas relacionadas con el metabolismo y almacenamiento del hierro, la ferritina y la transferrina.
CHCM ligeramente bajo
Los valores de CHCM ligeramente bajos (29-32 g/dl) se corresponden con la anemia ferropénica en el caso de que la hemoglobina también presente unos valores ligeramente disminuidos (9-10 g/dl).
CHCM moderadamente bajo
Los valores de CHCM moderadamente bajos (25-29 g/dl) se corresponden con anemias ferropénicas severas con hemoglobinemias muy reducidas.
CHCM muy bajo
Los valores de CHCM muy bajos (inferiores a 25 g/dl) se corresponden con anemias ferropénica muy severas. Suelen ser poco comunes.
¿Qué indican unos valores de CHCM altos?
Los valores altos de CHCM indican que la hemoglobina aparece muy concentrada en los glóbulos rojos.
Esta situación puede darse en circunstancias como haber sufrido grandes quemaduras corporales y en determinados tipos de anemias hemolíticas:
Esferocitosis . Se trata de una enfermedad hereditaria en la que los glóbulos rojos presentan una anomalía en la membrana que determina una forma esférica en lugar de la típica forma de disco. Provoca anemia hemolítica leve y se trata de la patología más común entre las que provocan unos valores aumentados de CHCM.
Drepanocitosis . Se trata de una patología hereditaria que cursa con anemia hemolítica crónica en la que los eritrocitos presentan una forma curvada o de hoz. Afecta casi exclusivamente a individuos de raza negra.
Xerocitosis congénita . Es una enfermedad hereditaria que provoca la rigidez de los glóbulos rojos, siendo una causa muy poco frecuente de anemia hemolítica.
Anemias hemolíticas autoinmunes . Tienen lugar cuando se forman anticuerpos que destruyen los glóbulos rojos del propio cuerpo. Suelen ser poco habituales.
Las patologías que suelen causar unos valores elevados de CHCM son enfermedades congénitas, cuyos valores son complicados de revertir.
CHCM ligeramente alto
Los valores de CHCM ligeramente altos (36-37 g/dl) se pueden corresponder con una esferocitosis o drepanocitosis.
CHCM muy alto
Los valores de CHCM muy altos (por encima 37 g/dl) suelen estar relacionados con problemas o errores en la medición y no con una patología real.

Artículo especializado
Cómo mejorar la circulación de forma rápida
El sistema circulatorio es el encargado de transportar el oxígeno y los nutrientes a todos los tejidos del organismo. Además, también contribuye al mantenimiento de la temperatura de forma estable en todas las partes del cuerpo. Su correcto funcionamiento es esencial para mantener una buena salud.
Contribuir a una buena circulación
En el correcto funcionamiento del sistema cardiovascular intervienen múltiples factores. Algunos de ellos no son controlables, como los factores hereditarios, pero existen hábitos y comportamientos que pueden influir de forma directa en un mejor estado del sistema circulatorio. Algunos de estos factores son:
Una adecuada alimentación . Como en muchos otros casos, la alimentación juega un papel fundamental en el correcto funcionamiento del sistema cardiovascular. La dieta debe ser rica en alimentos vegetales (verduras, frutas, legumbres, frutos secos, semillas y cereales integrales) y limitada en alimentos ricos en grasas saturadas (mantequilla, aceite de palma, carnes grasas, etc.), azúcares, derivados cárnicos y alimentos hiperprocesados y ricos en sal y aditivos.
Una hidratación suficiente . El agua forma parte fundamental de la sangre y la circulación. Beber lo suficiente, sobre todo agua, contribuirá a un mejor funcionamiento circulatorio.
Actividad física regular . El sedentarismo y la inactividad es uno de los principales factores de riesgo relacionados con una mala salud en general y una mala salud cardiovascular en particular. Realizar actividad física diaria y, siempre adaptada a las características de cada persona, es la mejor medida para optimizar el funcionamiento de nuestro sistema cardiovascular. Será positivo desde caminar 30 minutos al día hasta retos más ambiciosos.
Otras recomendaciones para mejorar el sistema cardiovascular pueden incluir la realización de masajes para reactivar la circulación. Una ducha fría, junto con un masaje ascendente en las extremidades inferiores al final del día es bueno para mejorar y reactivar la circulación venosa. Además, la utilización de ropa cómoda y holgada y zapatos planos también pueden beneficiar al retorno venoso y a tu bienestar.
Cómo mejorar la circulación en los pies
Para prevenir los problemas y mejorar la circulación en los pies y las piernas, algunos consejos incluyen:
Evitar permanecer de pie o sentado durante largos periodos. Es recomendable hacer movimientos, ejercicios o dar breves paseos de forma periódica para reactivar la circulación. Si se está sentado, es útil utilizar un reposapiés.
Elevar las piernas. Tras largos periodos sentados o de pie puede ser beneficioso para promover el retorno venoso tumbarse y elevar las piernas por encima del nivel del corazón.
El uso de medias terapéuticas puede aliviar molestias y la pesadez de las piernas favoreciendo la circulación de las extremidades inferiores.
Actividad física. Casi la totalidad de los deportes y actividades físicas ponen en movimiento las piernas (natación, correr, caminar, bicicleta, baile, gimnasia, etc.). Cualquier actividad es beneficiosa para la circulación de los pies.
Pies en remojo. Poner los pies en agua fría al final del día puede ser una manera sencilla y útil de reactivar la circulación en esa zona.
Calzado cómodo. Unos zapatos cómodos y sin tacón pueden ser beneficiosos para la circulación de las piernas y los pies.
Evitar el calor. La temperatura elevada dilata las venas y dificulta la circulación. Es importante intentar evitar las fuentes de calor directas a las piernas (depilación con cera caliente, uso de braseros, etc.).
Consultar con el médico ante cualquier molestia o problema circulatorio.
Cómo mejorar la circulación en las manos
Para prevenir los problemas y mejorar la circulación en las manos se pueden realizar algunos ejercicios, además de cuidar los factores generales para una buena circulación. Algunos de ellos pueden incluir:
Girar las muñecas hacia los dos lados. Realizar 30 repeticiones.
Abrir y cerrar las manos, estirando los dedos. Realizar 30 repeticiones.
Apretar las manos y relajarlas con la ayuda de una pelota antiestrés.
La práctica de ejercicio en general también contribuirá a una mejor circulación de las extremidades.
¿Qué empeora la circulación?
La mala circulación y su empeoramiento puede deberse a distintos factores, incluyendo:
Una alimentación deficiente , con un exceso de alimentos procesados, de origen animal, ricos en grasa saturadas, sal y azúcar, además de un consumo elevado de alcohol y un consumo insuficiente de frutas y verduras puede suponer un grave perjuicio para el sistema cardiovascular y para la circulación.
El tabaquismo es uno de los factores de riesgo más importantes para las enfermedades del sistema cardiovascular.
El sobrepeso puede dificultar el retorno venoso y empeorar la circulación. Además, el exceso de peso puede suponer un aumento del colesterol plasmático y de la tensión arterial, con el consiguiente aumento del riesgo cardiovascular.
El sedentarismo es otro de los factores que determinan un peor retorno venoso y un peor funcionamiento del corazón y el sistema vascular.
La inmovilidad física o permanecer mucho tiempo de pie o sentado dificulta retorno venoso de la sangre.
El embarazo es una etapa en la mujer en la que se produce un aumento significativo de peso y de líquido, por la formación del feto, la placenta, el líquido amniótico, etc. Esto, junto con las modificaciones hormonales, puede provocar una dificultad añadida para el sistema circulatorio de la mujer embarazada.
Los anticonceptivos orales pueden aumentar el riesgo de formación de trombos en algunas mujeres y empeorar el funcionamiento del sistema circulatorio.
La edad y el envejecimiento también son factores relacionados con una degradación de las estructuras vasculares y un peor funcionamiento de la circulación y el sistema cardiovascular.
Patologías crónicas como la hipertensión arterial, la diabetes mellitus y la obesidad pueden tener un impacto negativo sobre la circulación, además de ser factores de riesgo para los eventos cardiovasculares.
Síntomas de una mala circulación
Una mala circulación se puede hacer evidente a partir de ciertos síntomas, incluyendo:
Sensación de hormigueo. Puede estar motivado un bloqueo o un flujo deficiente de la circulación sanguínea.
Pesadez, hinchazón y edemas. Un mal retorno venoso puede provocar la acumulación de líquido, especialmente en las extremidades inferiores.
En casos extremos de problemas circulatorios pueden aparecer coloraciones azuladas o negras en las uñas de los pies, así como dificultad de movimiento, problemas para la cicatrización de heridas, etc.
Aparición de telangiectasias. Son las coloquialmente conocidas como “arañas vasculares”. Aparecen pequeñas formas ramificadas de color rojizo o azulado debajo de la piel, siendo un síntoma de una mala circulación.
La debilidad de las paredes venosas determina que la sangre se acumule y se formen las varices.
El cansancio y la pesadez en las piernas pueden ser indicativos de una mala circulación.
¿Puedo confiar en Savia?
Todos nuestros servicios están creados con la intención de acercar la atención médica y sanitaria de calidad a todos los trabajadores y sus familias, siempre con la garantía de MAPFRE. Queremos establecer una relación de total confianza.