Artículos Especializados

Artículo especializado
Consejos para tener una piel sana y bonita
La piel es el mayor órgano del cuerpo humano, cuyas principales funciones son proteger al organismo e impedir la entrada de microorganismos y sustancias potencialmente perjudiciales. Una piel sana se caracteriza por un funcionamiento adecuado, asociado a un buen aspecto.
Qué es una piel sana
Una piel sana es la consecuencia de un funcionamiento adecuado como órgano de excreción, sensitivo y estético. Esto incluye cumplir su funcionalidad como órgano barrera, de defensa inmunológica, activador de vitamina D, productor de melanina y regulador de temperatura.
Para ello, debe presentar todas sus estructuras en funcionamiento de manera adecuada, junto con la presencia de un microbioma equilibrado, es decir, la presencia de microorganismos beneficiosos para la piel adaptados a sus condiciones que impidan la aparición de especies patógenas.
De este modo, las características de una piel sana incluirían:
Una correcta hidratación de la piel.
Resistencia y elasticidad.
Tersura y brillo.
Ausencia de manchas y rojeces.
Sin descamación.
Sin exceso de grasa.
Ausencia de patologías de la piel como acné, dermatitis seborreica, dermatitis atópica, psoriasis o rosácea, entre otras.
¿Qué manchas y marcas afectan de forma significativa a la salud de la piel?
En la piel pueden aparecer diversos tipos de manchas que pueden revertir una gravedad variable.
Son especialmente relevantes las manchas que pueden estar asociadas al cáncer de piel. El melanoma es el tipo de cáncer de piel más grave, afectando a las células productoras de melanina. De este modo, los melanomas suelen ir asociados a la aparición de una mancha o lunar con unas características específicas. La técnica del “ ABCDE” es útil para saber si debemos realizar una consulta médica al observar un lunar o mancha de la piel:
A. Asimetría . Si se divide el lunar o la lesión en dos, cada mitad es diferente en forma y tamaño a la otra.
B. Borde irregular . Los bordes de un melanoma son irregulares, borrosos y poco definidos.
C. Cambiante . Se pueden producir cambios de color en la superficie del lunar. Cambia de forma o hay más de un color (marrón, negro, rojizo, etc.).
D. Diámetro . Un diámetro de más de medio centímetro de la lesión debe despertar sospechas.
E. Elevación . La lesión o el lunar cobran relieve.
Cuando alguno de los puntos se cumple, se recomienda acudir al Dermatólogo. Los melanomas suelen estar relacionados con una exposición excesiva a la radiación solar.
Por otro lado, dos de las manchas que suelen aparecer de forma más frecuente en la población y que también están relacionadas con la exposición a la luz solar, incluyen:
El lentigo senil o actínico . Se trata de una respuesta de la piel a la radiación ultravioleta. Afecta a zonas expuestas a la luz del sol como cara, cuello, escote y brazos. Son lesiones que aparecen normalmente a partir de los 60 años relacionadas con una historia de larga exposición solar y quemaduras, aunque pueden aparecer antes.
El melasma . Se trata de manchas irregulares en la cara, cuello y antebrazos. Son manchas marrones de mayor o menor intensidad. Suele aparecer en mujeres a partir de los 20 años, especialmente durante el embarazo o con la toma de anticonceptivos orales. La pigmentación de las manchas se incrementa durante el verano por la mayor exposición solar.
Este tipo de manchas no revisten gravedad, más allá de su afectación estética.
Otro tipo de manchas menos comunes que pueden aparecer en la piel pueden tener causas muy diversas. Entre ellas se incluyen:
Vitíligo. Se caracteriza por la aparición de grandes áreas de despigmentación causada por una enfermedad crónica de la piel de carácter autoinmune.
Dermatitis atópica. La dermatitis puede ser una causa de aparición de manchas en la piel.
Hipomelanosis. La hipomelanosis en gotas es un trastorno de la pigmentación caracterizado por la aparición de manchas blancas en la piel. Suelen ser manchas pequeñas y redondas que aparecen en las áreas más expuestas al sol.
Pitiriasis versicolor. Se trata de una infección superficial de la piel causada por hongos, en concreto por la proliferación de levaduras del género Malassezia, que forman parte de la flora habitual de la piel.
En cualquier caso, todas las manchas que parecen en la piel deben ser valoradas por el Dermatólogo.
Indicadores que definen una piel sana
Para identificar una piel sana es necesario analizar el funcionamiento de sus estructuras. Además, la presencia de los microorganismos adecuados impedirá la aparición de especies patógenas que pueden causar infecciones y el deterioro de zonas específicas de la piel. Otros elementos relevantes son la nutrición e hidratación adecuadas de la piel, tanto a nivel orgánico como de manera tópica. Finalmente, la higiene es fundamental para que la piel tenga salud y muestre un aspecto saludable.
De este modo, los indicadores o parámetros relacionados con una piel sana incluyen:
Ausencia de manchas en la piel, es decir, con un correcto funcionamiento de las células que producen el pigmento, los melanocitos y una correcta protección frente a la radiación solar.
Una piel sin exceso de grasa, con un funcionamiento correcto de las glándulas sebáceas.
Ausencia de rojeces o dilataciones vasculares en la piel, con una adecuada microcirculación de la piel.
Una piel sana es una piel hidratada, tolerante y resistente, es decir con una función barrera preservada.
Una piel libre de patologías. Incide aquí el conjunto de microorganismos y sus genomas que conviven en nuestra piel dando lugar a equilibrado ecosistema que permite el correcto funcionamiento de las estructuras cutáneas.
¿Qué consejos podrías dar para tener una piel saludable?
Para conseguir una piel sana es importante recalcar la importancia de potenciar estilos de vida saludables, incluyendo el control del estrés, ejercicio moderado y adecuado a la edad, alimentación saludable y equilibrada, abstención de tabaco y alcohol, hábitos sexuales sanos y hábitos solares saludables. También es importante controlar factores que influyan negativamente en la salud cutánea, como las agresiones ambientales (microorganismos, factores físicos, químicos, contaminantes, sol y radiaciones), y potenciar sistemas de asistencia sanitaria que permiten y favorezcan un adecuado control dermatológico.
Dentro de los cuidados para que tu piel esté sana y muestre su mejor aspecto se incluyen:
Hidratación e higiene. Limpiar la cara por la mañana y por la noche con un jabón adecuado al tipo de piel. Esto permite eliminar la suciedad y el exceso de grasa antes de aplicar cualquier cosmético.
Mantén la temperatura del agua del baño o ducha templada.
No utilizar cosméticos inadecuados o agresivos.
En el exterior no olvides la fotoprotección. Utiliza protector solar de amplio espectro frente a UVB, UVA y luz visible durante todo el año.
Elige prendas de vestir que eviten la sudoración (transpirables, holgadas, de tejidos naturales).
Visita al dermatólogo al menos una vez al año para detectar todas las alteraciones de la piel y corregirlas mediante un tratamiento dermatológico para conseguir una piel sana y como consecuencia una piel bonita.
Elije bien tus productos
Para mantener una piel saludable y con buen aspecto es esencial utilizar los productos adecuados:
Fórmulas hidratantes y emolientes. La aplicación de sustancias y fórmulas emolientes pueden ser útiles en todas aquellas situaciones en las que la barrera cutánea está alterada. Los emolientes son sustancias de uso externo capaces de aumentar la hidratación, promoviendo la formación de una película que limita la evaporación del agua. Incluyen sustancias como glicerina, siliconas, aceites vegetales, entre otras.
Usa un transformador de la piel, idealmente asesorado por el dermatólogo ya que no todas las pieles ni en todos los momentos del año necesitan lo mismo. El ácido glicólico y el retinol son los principios activos estrella, permitiendo corregir pequeñas manchas y arrugas. También mejoran la hidratación de la piel y aportan resistencia y luminosidad a largo plazo.
Las ceramidas también pueden contribuir a reestablecer la función barrera de la piel.
No renuncies a la calidad.
Ante cualquier afección y para mantener una piel sana y bonita puedes consultar con la especialista en Dermatología Paula Aguayo Carreras aquí .
Dra. Paula Aguayo Carreras
Especialista en Medicina Estética
Dermatóloga experta en Dermatología Estética. Dermatóloga asesora de la marca La Roche-Posay (2020- actualidad). Actualmente trabajo en la Clínica Dermatológica y Estética Dermacenter de Málaga y en el Hospital Regional Universitario de Málaga. Doctora en Medicina y Cirugía por la Universidad de Granada (noviembre 2021). Premio al Mejor Expediente MIR de Andalucía 2019 por el Consejo Andaluz de Colegio de Médicos.
Reserva ya tu videollamada
Bibliografía
Academia Española de Dermatología y Venereología. Fundación Piel Sana. ¿Cuándo está la piel sana? 03/02/2016. Consultado: 16/01/2023.
Harris-Tryon TA, Grice EA. Microbiota and maintenance of skin barrier function. Science 2022; 376(6596): 940-945. Doi: 10.1126/science.abo0693.
Academia Española de Dermatología y Venereología. El DNI de las manchas. Consultado: 16/01/2023.
David Boothe W, Tarbox JA, Tarbox MB. Atopic Dermatitis: Pathophysiology. Adv Exp Med Biol 2017; 1027: 21-37. Doi: 10.1007/978-3-319-64804-0_3.

Artículo especializado
Resultados de test prenatales: ¿Cuánto tardan y cómo leerlos?
El test prenatal no invasivo es una prueba que permite comprobar el estado del bebé antes de su nacimiento e identificar posibles anomalías genéticas, así como conocer el sexo fetal. Esta prueba se realiza a partir de una muestra de sangre de la madre y sus resultados están disponibles en unos días.
Test prenatales no invasivos
La técnica de los test prenatales se basa en analizar el ADN fetal que está presente en la sangre de la madre durante el embarazo. De este modo, a partir de una muestra de sangre materna se analizan las posibles anomalías cromosómicas del feto en gestación.
La información genética se organiza dentro de cada célula en los cromosomas. Los seres humanos tenemos 23 pares de cromosomas (dos copias de cada par, es decir 46). Los problemas de salud y de desarrollo aparecen cuando falta o sobra algún cromosoma o una parte de ellos.
En este sentido, los test emplean la última tecnología en secuenciación que compara el ADN fetal respecto al ADN materno para detectar determinadas anomalías con gran precisión y fiabilidad. Además, pueden incluir otras pruebas para la detección de trisomías relacionadas con abortos espontáneos en el primer trimestre e identifican microdeleciones en las que una pérdida de material genético puede causar la aparición de síndromes genéticos importantes.
Trisomías
En las trisomías se produce la presencia de un cromosoma adicional, es decir, 3 copias de un cromosoma en lugar de las 2 copias normales.
Los defectos cromosómicos más habituales en forma de trisomías que pueden detectar los test prenatales son los que aparecen en los cromosomas 21, 18 y 13 (síndrome de Down, Edwards y Patau) y las anomalías en los cromosomas sexuales (X e Y).
Microdeleciones
Las microdeleciones son pequeñas pérdidas de material genético que se asocian con graves problemas del desarrollo en el recién nacido. Se incluyen aquí el síndrome de DiGeorge, el síndrome de deleción 1p36, el síndrome de Cri-du-chat, el síndrome de Angelman y el síndrome de Prader-Willi.
El síndrome por microdeleción más frecuente es el de DiGeorge. En él está ausente un pequeño fragmento del cromosoma 22 (22q11.2), lo que acarrea múltiples alteraciones fetales como defectos cardíacos, anomalías en el paladar y la cara, retraso en el desarrollo e inmunodeficiencia, con infecciones frecuentes.
Triploidía
La triploidía es una alteración cromosómica en la que el feto tiene tres copias de cada uno de los cromosomas en lugar de dos (69 cromosomas en total y no 46). Normalmente se produce cuando dos espermatozoides fertilizan un óvulo, pero también puede ocurrir cuando un óvulo diploide es fertilizado por un espermatozoide, o bien en casos en los que un espermatozoide diploide fertiliza un óvulo.
¿Cómo interpretar resultados test prenatal no invasivo?
El Test Prenatal No Invasivo se basa en tecnología de ultrasecuenciación o Next Generation DNA Sequencing , junto con herramientas avanzadas de bioinformática. El test cuantifica la probabilidad de que esté presente alguna de las alteraciones genéticas más comunes. El resultado ofrece una especificidad y sensibilidad muy elevadas.
La interpretación de los resultados se realiza en función de la estimación del riesgo:
Bajo riesgo . Existe una probabilidad de más del 99% de que el bebé no presente ninguna de las anomalías cromosómicas analizadas. Sin embargo, no puede descartar por completo la posibilidad de afectación cromosómica fetal.
Alto riesgo . la probabilidad de alteración cromosómica o de microdeleción es elevada. En esta situación el ginecólogo recomendará la realización de una prueba de diagnóstico prenatal invasiva para confirmar el diagnóstico.
El resultado viene expresado en porcentaje de riesgo, que oscila entre 0,01 y 99%.
En definitiva, a pesar de su alta fiabilidad, se trata de un test de cribado, por lo que, en caso de resultado positivo, este debe ser confirmado mediante una prueba invasiva (amniocentesis o biopsia corial).
¿Qué es capaz de detectar el test prenatal?
Trisomías. En lugar de dos cromosomas, uno de la madre y otro del padre, hay un tercero. Dependiendo del cromosoma que se trate, puede ser:
Trisomía 21. Síndrome de Down.
Trisomía 18. Síndrome de Edwards.
Trisomía 13. Síndrome de Patau.
Anomalías en los cromosomas sexuales:
Monosomía X (un solo cromosoma X). Síndrome de Turner.
Múltiples cromosomas Y. Síndrome de Klinefelter.
Están ausentes pequeños fragmentos cromosómicos.
Está ausente un fragmento del cromosoma 22 (22q11.2). Síndrome de DiGeorge.
Otras microdeleciones que dan lugar al síndrome de deleción 1p36, el síndrome de Cri-du-chat, el síndrome de Angelman y el síndrome de Prader-Willi.
Triploidía.
Sexo fetal.
¿Cuánto tardan los resultados de un test no invasivo?
Los resultados de los test no invasivos están basados en el análisis del ADN fetal presente en la sangre materna. Estos pueden estar disponibles, aproximadamente, en una semana.
¿Qué fiabilidad tienen los test prenatales no invasivos?
La fiabilidad del test prenatal no invasivo es muy elevada, situándose por encima del 99% respecto a la detección de las trisomías más habituales (síndromes de Down, Patau y Edwards y las que afectan a los cromosomas sexuales).
La tasa de falsos positivos se sitúa en menos del 0,1%. De igual modo, la tasa de falsos negativos en el test prenatal es muy baja y se relaciona con niveles de ADN fetal muy bajo en la sangre materna.
Limitaciones del test prenatal
Algunas situaciones pueden limitar la utilidad del test, por lo que no se recomienda su uso. Entre ellas se encuentran:
Embarazo inferior a 9 semanas en el momento de la prueba.
Embarazo de 3 o más fetos o con constancia de gemelo evanescente.
Embarazo mediante donación ovular. En este caso el riesgo de que el bebé presente alguna alteración cromosómica suele ser menor, dado que estos óvulos suelen proceder de mujeres jóvenes y correctamente estudiadas. Pero si fuera necesario, se podría realizar con la misma eficacia diagnóstica que en fetos procedentes de óvulos propios de la madre.
Haber recibido recientemente una transfusión sanguínea, trasplante de médula ósea o terapia de células madre.
Los mosaicismos fetales de las trisomías, así como alteraciones parciales de los cromosomas estudiados, pueden no ser detectados.
A pesar de su alta fiabilidad, se trata de un test de cribado, por lo que, en caso de resultado positivo, éste debe ser confirmado mediante una prueba invasiva (amniocentesis o biopsia corial). En cualquier caso, la realización del test prenatal está exenta de riesgo para el feto y para la madre.
Artículo especializado
Microdeleción: ¿Qué es y qué síndromes genera?
La ausencia de una parte de material genético en los cromosomas puede determinar la aparición de diversos síndromes congénitos.
¿Qué son las microdeleciones?
Las microdeleciones son anomalías cromosómicas causadas por pequeñas pérdidas de material genético que aparecen de forma congénita y que están presentes durante la gestación. Estas ausencias de genes determinan la aparición de graves síndromes que determinan problemas del desarrollo en el recién nacido.
Los diferentes síndromes dependen del cromosoma y la zona específica en la que se ha producido la pérdida genética. Estos tienen características clínicas distintivas que permiten muchas veces sospechar la microdeleción subyacente.
Cromosomas e información genética
La información genética presente en el ADN se organiza dentro de cada célula en los cromosomas. Los seres humanos tenemos 23 pares de cromosomas (dos copias de cada par, es decir 46). Los primeros 22 pares se numeran del 1 al 22. El último par determina el sexo. Las niñas tienen dos cromosomas X y los niños un cromosoma X y un cromosoma Y. Los problemas de salud y de desarrollo aparecen cuando falta (microdeleciones) o sobra algún cromosoma o una parte de ellos.
La mayoría de las deleciones cromosómicas ocurren como un acontecimiento al azar durante la formación de las células reproductoras (óvulos o espermatozoides) o durante el desarrollo fetal temprano. De este modo, no existen fatores hereditarios y los padres presentan cromosomas normales. En estas microdeleciones la edad materna tampoco influye.
Las anomalías cromosómicas, incluyendo las microdeleciones más comunes, pueden ser detectadas gracias a los test prenatales no invasivos , sin riesgo para la madre ni el bebé.
¿Cuáles son los síndromes de microdeleción?
Existen diferentes síndromes causados por microdeleción con características clínicas diferenciadas. Entre los más comunes se encuentran:
Síndrome de Smith-Magenis
El Síndrome de Smith-Magenis tiene una incidencia cercana a 1 caso por cada 25.000 recién nacidos.
Corresponde a una deleción en el cromosoma 17, en concreto la banda p11.2 (17p11.2).
Se caracteriza por múltiples anomalías congénitas, junto con retraso mental. El fenotipo característico incluye los siguientes signos:
Braquicefalia o aplanamiento de la parte posterior de la cabeza.
Hipoplasia mediofacial, con desarrollo insuficiente de los huesos maxilares.
Mandíbula prominente.
Voz ronca.
Alteraciones del sueño.
Síndrome de Prader-Willi
El Síndrome de Prader-Willi afecta a 1 de cada 15.000-30.000 recién nacidos.
Está causado por la ausencia de contribución paterna de una región en el cromosoma 15 (en concreto la 15q11.2q11.3).
Se caracteriza por la presencia de:
Déficit cognitivo y trastorno de conducta.
Hipotonía severa.
Dificultad para alimentarse durante la lactancia.
Hiperfagia entre 1 y 6 años de edad con desarrollo gradual de obesidad mórbida.
Hipogonadismo en ambos sexos.
Talla baja.
Síndrome de Angelman
Este síndrome tiene una incidencia 1 caso entre cada 10.000-20.000 nacimientos.
Está causado por la ausencia de la contribución materna de la región del cromosoma 15 (en concreto el gen UBE3A).
Se caracteriza, en más de 90% de los pacientes, por una historia prenatal y de recién nacido normal con un perfil metabólico normal y un sistema nervioso central estructuralmente normal, pero con la posterior presencia de:
Retraso mental severo.
Ausencia de lenguaje.
Ataxia, temblor de las extremidades y convulsiones.
Microcefalia.
Estrabismo.
Hipopigmentación de piel y ojos.
Protrusión lingual con alteración de la succión-deglución y problemas de alimentación.
Boca amplia con dientes separados y mandíbula prominente.
Brazos flectados en la marcha.
Hipersensibilidad al calor.
Atracción por el agua.
Alteraciones del sueño.
Síndrome de DiGeorge
El síndrome de DiGeorge es el síndrome por microdeleción más común. Presenta una incidencia de uno de cada 2.000-4.000 recién nacidos vivos.
Se trata de un cuadro con expresión variable, causado por una deleción submicroscópica en el cromosoma 22 (22q11.2) en el 95 % de los casos. Las personas afectadas también presentan un mayor riesgo de autismo, trastorno de déficit de atención y otras afecciones sociales y neurológicas.
Cursa con:
Cardiopatía.
Anomalías del paladar.
Hipocalcemia.
Inmunodeficiencia.
Trastorno del aprendizaje.
Facies característica.
Síndrome de Cri-Du-Chat
El síndrome de “cri du chat” o “maullido de gato” es una enfermedad hereditaria rara que afecta a 1 de cada 20.000-50.000 recién nacidos.
Está causado por la deleción de material genético en uno de los brazos del cromosoma 5.
Se caracteriza por el hecho de que el bebé lactante afectado presenta un llanto similar al maullido de un gato.
Se caracteriza, además, por la presencia de los siguientes signos clínicos:
Anomalías cardíacas
Discapacidad intelectual.
Bajo peso al nacer y crecimiento lento.
Retraso de las habilidades motoras.
Pérdida de la audición.
Ojos separados e inclinados hacia abajo.
Aparición de un pliegue adicional de piel sobre el ángulo interior del ojo.
Orejas de implantación baja o de forma anormal.
Fusión o formación parcial de membranas en los dedos de las manos o los pies.
Escoliosis.
Una sola línea en la palma de la mano.
Microcefalia.
Síndrome de Deleción 1p36
El síndrome de deleción 1p36 es una anomalía cromosómica que puede afectar a 1 de cada 5.000 nacidos vivos.
El síndrome de deleción 1p36 está causado por la ausencia genética que afecta principalmente al extremo terminal del brazo corto del cromosoma 1.
Los signos distintivos del síndrome incluyen:
Rasgos faciales dismórficos (microbraquicefalia, una fontanela anterior amplia con retraso de cierre, cejas rectas, ojos hundidos, puente nasal ancho y deprimido, protrusión del tercio medio facial, orejas anómalas de implantación baja y rotadas posteriormente, surco subnasal largo y barbilla puntiaguda).
Dedos y pies cortos.
Hipotonía.
Retraso del desarrollo.
Discapacidad intelectual.
Crisis epilépticas.
Defectos cardíacos.
Alteración o ausencia del habla.
Restricción del crecimiento de inicio prenatal.
Anomalías esqueléticas.
Anomalías en los genitales.
Síndrome de Wolf-Hirschhorn
El síndrome de Wolf-Hirschhorn es un trastorno genético que afecta a 1 de cada 20.000-50.000 recién nacidos. Es más frecuente en mujeres.
Está causado por la deleción de material genético cerca del extremo del brazo corto del cromosoma 4. El tamaño de la deleción varía, mostrándose síntomas más graves en las deleciones mayores.
Se caracteriza por la presencia de los siguientes signos:
Apariencia facial característica (microcefalia, nariz ancha, frente alta con glabela prominente, ojos separados, cejas arqueadas y altas, surco nasolabial corto, boca curvada hacia abajo, orejas malformadas y mandíbula muy pequeña).
Retraso del crecimiento y del desarrollo.
Discapacidad intelectual.
Bajo tono muscular (hipotonía) y convulsiones.
Anomalías de los huesos.
Defectos congénitos cardíacos.
Pérdida de la audición.
Malformaciones del tracto urinario.
Anormalidades estructurales del cerebro.
Síndrome de Jacobsen
El síndrome de Jacobsen es una cromosomopatía infrecuente que afecta a uno de cada 100.000 nacimientos.
Está causado por una deleción terminal del brazo largo del cromosoma 11.
Los principales rasgos clínicos incluyen:
Retraso psicomotor y del crecimiento.
Trigonocefalia (forma triangular o en cuña de la frente).
Dismorfia facial característica.
Hematomas y petequias generalizadas.
Pies zambos.
Anomalías digestivas y renales.
Síndrome de Langer-Giedion
El síndrome de Langer-Giedion es un síndrome muy poco frecuente que afecta a menos de un caso por cada millón de nacimientos.
Está causado por una deleción del cromosoma 8 (8q 23.3-q24.11) que afecta a los genes EXT 1 y TRPS 1.
El espectro clínico incluye:
Retraso mental.
Talla baja.
Microcefalia.
Dismorfismo facial.
Exóstosis o formaciones de tejido óseo en el conducto auditivo.
Anormalidades esqueléticas.
Microdeleciones del cromosoma Y e infertilidad masculina
La infertilidad masculina puede tener su origen en causas genéticas. De este modo, las microdeleciones del cromosoma Y (cromosoma sexual masculino) pueden suponer una causa de infertilidad en el hombre.
El cromosoma Y posee una región denominada AZF o factor de azoospermia. Las deleciones en esta región cromosómica se pueden traducir en trastornos en la producción de esperma. Estos pueden incluir azoospermia o imposibilidad total de producir espermatozoides y oligospermia, en la que la producción de espermatozoides está disminuida.
¿Cuál es el síndrome de microdeleción más común?
El síndrome por microdeleción más frecuente es el síndrome de DiGeorge. En él está ausente un pequeño fragmento del cromosoma 22 (22q11.2), lo que acarrea múltiples alteraciones fetales como defectos cardíacos, anomalías en el paladar y la cara, retraso en el desarrollo e inmunodeficiencia, con infecciones frecuentes.
El asesoramiento genético ofrecido a través de los resultados de los test prenatales no invasivos ayuda a diagnosticar estas microdeleciones, favoreciendo el diagnóstico precoz y ofreciendo las opciones para su tratamiento y manejo oportunos.
Artículo especializado
Grupo sanguíneo en el embarazo: ¿Cuál trae problemas?
Todas las personas tienen un grupo sanguíneo (O, A, B o AB) y un factor Rh (positivo o negativo). El grupo sanguíneo y el factor Rh significan simplemente que la sangre de una persona tiene ciertas características específicas. El grupo sanguíneo se traduce en forma de proteínas en los glóbulos rojos y en los líquidos corporales.
Por otro lado, el factor Rh se identifica con una proteína que recubre los glóbulos rojos. Si esta proteína está presente, la persona tiene un factor Rh positivo. En cambio, si la proteína está ausente, la persona tiene factor Rh negativo.
Los factores Rh se determinan genéticamente. Un bebé puede tener el grupo sanguíneo y el factor Rh de cualquiera de sus padres o una combinación de ambos. Los factores Rh siguen un patrón común de herencia genética. El gen Rh positivo es dominante (más fuerte) e incluso cuando se combina con un gen Rh negativo, el positivo prevalece.
El problema tiene lugar cuando se ponen en contacto dos grupos sanguíneos con distintos factores Rh, provocando una respuesta inmunitaria. Esto puede suceder en una transfusión sanguínea, o cuando la madre embarazada es factor Rh positivo y el embrión en gestación es Rh negativo.
Estas manifestaciones se traducirán en una serie de eventos clínicos de distinta gravedad, según la cantidad sanguínea expuesta al contacto.
Grupo sanguíneo: sistema AB0
El grupo sanguíneo se basa en la presencia o ausencia de ciertos antígenos que se encuentran en la superficie de los glóbulos rojos. En este sentido, también son relevantes los anticuerpos que circulan en la sangre que reconocen estas células como propias.
Los glóbulos rojos pueden clasificarse en tres tipos: A, B y 0, de acuerdo con la presencia o ausencia de antígenos en la superficie.
Grupo A . Los glóbulos rojos de los individuos del grupo A tienen antígenos del tipo A en su superficie y anticuerpos anti-B en la sangre. Se estima que este grupo puede alcanzar a un tercio de la población.
Grupo B . Los glóbulos rojos presentan en su superficie antígenos tipo B y anticuerpos anti-A en la sangre. Cerca del 20% de la población es del grupo B.
Grupo AB . Los antígenos que aparecen en la superficie de los glóbulos rojos son de los dos tipos: A y B y no presentan anticuerpos en el plasma sanguíneo. Es el grupo sanguíneo menos común, alcanzando tan solo al 5% de la población.
Grupo 0. Los glóbulos rojos no presentan ningún antígeno en su superficie, por lo que tienen anticuerpos anti-A y anti-B en el plasma sanguíneo. Es el grupo mayoritario, con más del 40 % de la población.
¿Qué tipo de sangre tiene problemas en el embarazo?
Como se ha dicho, cuando un glóbulo rojo de un grupo sanguíneo entra en contacto con la sangre de otro grupo (transfusión o embarazo) provoca una reacción inmunológica en el huésped. En el caso de que exista una diferencia de grupo sanguíneo (A, B, AB y 0) entre la madre y el feto en gestación, no suelen aparecer problemas de consideración. De este modo, no hay afectación prenatal y la postnatal es leve o moderada.
Esta situación es debida a que los anticuerpos del sistema AB0 no son capaces de atravesar la placenta o lo hacen en cantidades muy limitadas, ya que se tratan del tipo IgM.
La incompatibilidad del factor Rh es la situación que tiene mayor importancia, ya que los anticuerpos anti-D son de clase IgG, capaces de atravesar la placenta, pudiendo provocar la enfermedad hemolítica del recién nacido. En esta los glóbulos rojos del feto son atacados por los anticuerpos maternos, ya que los reconoce como extraños y se pone en marcha un mecanismo para su destrucción.
Esta situación sucede cuando una mujer Rh negativa se queda embarazada, siendo el bebé Rh positivo. En este caso, se puede producir un intercambio entre la sangre del bebé y la de la madre durante el embarazo y, especialmente, durante el parto.
Cuando se trata del primer embarazo esta situación no suele ser peligrosa. Al contrario de lo que ocurre con el sistema AB0, los anticuerpos anti-Rh no se encuentran en el plasma de los individuos Rh negativo hasta que las células del sistema circulatorio de estos individuos hayan estado en contacto con el antígeno Rh positivo con anterioridad.
De este modo, tras ese primer embarazo, la madre tendrá anticuerpos frente al antígeno Rh positivo. En un segundo embarazo con las mismas circunstancias se produciría el ataque de los anticuerpos de la madre frente a los antígenos de los glóbulos rojos del bebé, provocando su hemólisis.
Enfermedad hemolítica del recién nacido
La enfermedad hemolítica del recién nacido es el proceso en el que se produce la destrucción acelerada de los glóbulos rojos fetales por parte de los anticuerpos maternos, generando una anemia grave en el bebé.
El intento de generación de nuevos hematíes para suplir esa carencia determina la presencia de eritroblastos, glóbulos rojos inmaduros incapaces de cumplir su función de forma correcta.
La destrucción masiva de hematíes produce una sustancia de deshecho, la bilirrubina, que a partir de ciertos niveles es tóxica. En primera instancia provoca ictericia, pero en casos graves, cuando la concentración va en aumento y se acumula, puede provocar trastornos neurológicos e incluso la muerte.
¿Qué pasa cuando una mujer embarazada es Rh negativo?
Como se ha dicho, cuando la madre es Rh negativo y el feto Rh positivo, esta puede generar anticuerpos frente al Rh de su hijo.
Esta reacción puede llegar a eliminar los glóbulos rojos del bebé, provocando anemia en el momento del parto y también a lo largo del embarazo.
Para evitar esta situación, en madres con Rh negativo se lleva a cabo el test de Coombs indirecto, una prueba que detecta anticuerpos frente al Rh positivo del feto capaces de atravesar la placenta, causando la enfermedad hemolítica del recién nacido.
En esos casos, se administra a la madre de forma preventiva en torno a las 28 semanas de gestación una gammaglobulina anti-D que impide la producción de anticuerpos contra las células sanguíneas del bebé.
Tras el parto, si se confirma que el Rh del recién nacido es positivo, durante las primeras 72 horas, habrá que repetir la dosis de gammaglobulina a la madre.
Después del embarazo, se pueden realizar transfusiones si el bebé padece de una anemia grave y administrar líquidos intravenosos si la tensión arterial es baja. En los casos en los que el bebé tiene ictericia, existe un procedimiento llamado exanguinotransfusión en el que se extrae sangre del bebé con un nivel alto de bilirrubina y se reemplaza por sangre nueva con un nivel normal. Además de bajar el nivel de bilirrubina, se aumenta de esta forma el recuento de glóbulos rojos. En los casos más leves de bilirrubinemia, está indicada la fototerapia para ayudar al bebé a eliminar el exceso de bilirrubina. También existe la opción de administrar Inmunoglobulina intravenosa para ayudar al sistema inmune del bebé y reducir la hemólisis de los glóbulos rojos.

Artículo especializado
Taquipnea: Qué es, causas y valores
La respiración es el proceso fisiológico vital por medio del cual se asegura el aporte necesario de oxígeno a los órganos y tejidos, así como se produce la eliminación del dióxido de carbono resultado del metabolismo celular.
El ritmo de respiración puede variar en función de diferentes factores, como la edad, el tipo de actividad que se está realizando o una enfermedad. Cuando la respiración está acelerada por encima de los niveles normales recibe el nombre de taquipnea.
La frecuencia respiratoria es un signo vital importante y en ocasiones la manifestación inicial de algunas enfermedades.
¿Qué es la taquipnea?
Taquipnea es el término médico que indica que la frecuencia respiratoria está por encima de los valores normales para su edad. Las situaciones que pueden causar taquipnea son muy variadas y, en muchos casos, están condicionadas por la edad y otras características de la persona.
Valores normales de la taquipnea en niños y adultos
Los valores de la frecuencia respiratoria son muy variables y se pueden modificar por diferentes circunstancias como el ejercicio, el estrés, la edad, estar situado en grandes alturas, la fiebre, el tratamiento con determinados medicamentos, las circunstancias del ambiente y ciertas enfermedades.
Los valores normales de número de respiraciones por minuto (frecuencia respiratoria) van a depender principalmente de la edad de la persona. Según se desarrolla el sistema respiratorio la frecuencia respiratoria va disminuyendo. Siendo mayor en los recién nacidos y menor en adultos. La frecuencia respiratoria normal en reposo en las diferentes franjas de edad se sitúa en:
Lactante (menos de 1 año): de 30 a 60 respiraciones por minuto.
Niño (de 1 a 3 años): de 24 a 40 respiraciones por minuto.
Preescolar (de 3 a 6 años): de 22 a 34 respiraciones por minuto.
Edad escolar (de 6 a 12 años): de 18 a 30 respiraciones por minuto.
Adolescencia (de 13 a 18 años): 12 a 15 respiraciones por minuto.
Adulto: se sitúa en torno a 10-20 respiraciones por minuto.
Causas de la taquipnea
La taquipnea puede estar causada por múltiples situaciones:
Fisiológicas: miedo, ansiedad, llanto, ejercicio, fiebre y dolor.
Patológicos.
Respiratorios:
Falta de oxígeno.
Aumento de dióxido de carbono.
Traumatismo
Edema pulmonar.
Tromboembolismo pulmonar.
Hipertensión pulmonar.
Derrame pleural.
NO respiratorios:
SNC: tumores e infecciones.
Cardiovascular: insuficiencia cardiaca, hipotensión arterial.
Metabólico: acidosis, insuficiencia hepática.
Fármacos.
Hematológico: anemia.
Psicógenos: ansiedad, estrés.
Entre las enfermedades respiratorias que producen taquipneas se encuentran:
Asma . Es una enfermedad caracterizada por una inflamación crónica de las vías respiratorias, que cursa con la aparición de síntomas como sibilancias (pitidos en los pulmones), dificultad para respirar, opresión en el pecho y tos. Con el asma se produce limitación del flujo aéreo respiratorio.
Tromboembolimo pulmonar . Se produce obstrucción de una parte del territorio arterial pulmonar (vasos sanguíneos que llevan sangre baja en oxígeno desde el corazón hasta los pulmones para oxigenarla) a causa de un trombo que procede de otra parte del cuerpo.
Enfermedad pulmonar obstructiva crónica (EPOC) . Esta patología crónica pulmonar causa la obstrucción crónica del flujo de aire en los pulmones. Se manifiesta con tos crónica y dificultad respiratoria. Su causa principal es el consumo de tabaco.
Otras enfermedades pulmonares graves, como enfermedad pulmonar intersticial, bronquiectasia, neumotórax o neumonía.
Bronquiolitis en bebés . La infección de los bronquiolos, las vías respiratorias más estrechas de los pulmones, propicia una aceleración del ritmo respiratorio.
La taquipnea transitoria del recién nacido . Tiene lugar cuando un recién nacido intenta expulsar los líquidos que se albergaban en sus pulmones durante el periodo de gestación. No se trata de una condición grave y suele resolverse espontáneamente en 2 o 3 días. Se caracteriza por un cuadro de dificultad respiratoria presente desde el nacimiento o 2 horas después. La respiración del bebe está acelerada pudiendo llegar a 100-120 respiraciones por minuto.
Síntomas habituales de la taquipnea
Los síntomas que suele acompañar a la taquipnea dependerán del motivo o la patología que los cause y pueden ser:
Disnea o dificultad respiratoria.
Tos y expectoración.
Dolor y opresión en el tórax.
Inestabilidad hemodinámica (en el embolismo pulmonar).
En la taquipnea transitoria del recién nacido puede aparecer cianosis (coloración azulada de la piel por la falta de oxígeno y exceso de CO 2 ), respiración acelerada con la emisión de sonidos que se asemejan a gruñidos, aleteo nasal y retracción intercostal.
Bradipnea
Al contrario de la taquipnea, la bradipnea es el término médico utilizado para referirse a la disminución de la frecuencia respiratoria. Suele estar definida por situaciones en las que se producen menos de 12 respiraciones por minuto en adultos. Existen diferentes causas de bradipnea como consumo de sustancias tóxicas, traumatismo costales, consumo de algunos fármacos, parálisis musculas.
Tratamiento de la taquipnea
El tratamiento de la taquipnea dependerá de las causas. Para mejorar la función respiratoria existen métodos terapéuticos como las sesiones de rehabilitación respiratoria. Si existe una patología respiratoria lo habitual es acudir al neumólogo para completar el estudio del funcionamiento respiratorio.

Artículo especializado
Síndrome de Smith-Magenis: ¿Qué es y cómo detectarlo?
Las microdeleciones son anomalías cromosómicas causadas por pequeñas pérdidas de material genético que pueden determinar la aparición de síndromes congénitos que cursan con problemas en el desarrollo del recién nacido.
Los diferentes síndromes dependen del cromosoma y la zona específica en la que se ha producido la pérdida genética. Uno de ellos es el Síndrome de Smith-Magenis.
¿Qué es el síndrome de Smith-Magenis?
El síndrome de Smith-Magenis está causado por una deleción o pérdida de material genético en el cromosoma 17 (banda G11.2p). Recientemente se ha comprobado que la pérdida o las mutaciones del gen RA/1 situado en esa región cromosómica podría ser el causante específico del síndrome.
El síndrome queda definido por un conjunto de características físicas y de conducta que incluyen retraso mental.
Se estima que el síndrome de Smith-Magenis afecta a uno de cada 25.000 recién nacidos (niños y niñas), si bien la incidencia podría ser mayor.
En la mayoría de los casos, la deleción de material genético tiene lugar de forma accidental, por lo que se puede calificar como un síndrome congénito, pero no hereditario.
Cromosomas e información genética
La información genética presente en el ADN se organiza dentro de cada célula en los cromosomas. Los seres humanos tenemos 23 pares de cromosomas (dos copias de cada par, es decir 46). Los primeros 22 pares se numeran del 1 al 22. El último par determina el sexo. Las niñas tienen dos cromosomas X y los niños un cromosoma X y un cromosoma Y. Los problemas de salud y de desarrollo aparecen cuando falta (microdeleciones) o sobra algún cromosoma o una parte de ellos. En el caso del síndrome de Smith-Magenis, el cromosoma afectado es el 17.
La mayoría de las deleciones cromosómicas ocurren como un acontecimiento al azar durante la formación de las células reproductoras (óvulos o espermatozoides) o durante el desarrollo fetal temprano. De este modo, no existen fatores hereditarios y los padres suelen presentar cromosomas normales. En estas microdeleciones la edad materna tampoco influye.
Las anomalías cromosómicas, incluyendo las microdeleciones más comunes, pueden ser detectadas gracias a los test prenatales no invasivos , sin riesgo para la madre ni el bebé.
Síndrome de Smith-Magenis: caracteristicas
Los afectados por el síndrome de Smith-Magenis presentan una gran variedad de signos y síntomas que se hacen más evidentes con la edad.
El nacimiento del bebé suele ser a término y con unas características normales. Posteriormente empiezan a aparecer hipotonía, letargia y problemas en la alimentación, lo que determina una ralentización del crecimiento.
El fenotipo característico del síndrome de Smith-Magenis incluye los siguientes signos:
Características físicas y fisiológicas
Braquicefalia o aplanamiento de la parte posterior de la cabeza.
Hipoplasia mediofacial, con desarrollo insuficiente de los huesos maxilares.
Mandíbula prominente.
Voz ronca.
Manos y pies cortos y anchos.
Talla baja.
Escoliosis.
Anomalías en el oído medio.
Inestabilidad al caminar.
Piel áspera.
Hipotonía.
Anomalías renales o urinarias.
Alteraciones cardíacas.
Trastornos tiroideos.
Desprendimiento de retina.
Estreñimiento.
Características cognitivas y de comportamiento
Retraso mental.
Alteraciones del sueño.
Retraso del lenguaje.
Problemas para aprender a leer.
Estereotipias o movimientos repetitivos y convulsiones.
Conductas autoagresivas.
Diagnóstico
Con un test prenatal no invasivo se puede detectar la mayoría de las deleciones 17p11.2, sin ningún riesgo para la madre ni el bebé, a partir del material genético del feto presente en la sangre materna. Otras alternativas son las pruebas prenatales invasivas como la amniocentesis o la biopsia corial.
Con posterioridad al nacimiento, el diagnóstico precoz del síndrome es de gran ayuda a los padres para aprender las características de la enfermedad y las necesidades específicas de los niños afectados.
Este síndrome por microdeleción comparte características y signos con otros síndromes y patologías, de ahí las dificultades para establecer un diagnóstico clínico. Entre ellos se cuentan el síndrome de Prader-Willi (deleción en el cromosoma 15), el síndrome de Williams (deleción en el cromosoma 7), los trastornos del espectro autista y el trastorno por déficit de atención e hiperactividad (TDAH).
Tratamiento
El síndrome de Smith-Magenis, como el resto de los síndromes cromosómicos congénitos no se puede curar. Sin embargo, muchos de los signos y características del síndrome pueden mejorar con diferentes tratamientos establecidos por un equipo médico multidisciplinar. Algunos de estos tratamientos incluyen, entre otros:
Medicación psicotrópica para estabilizar los cambios del humor, disminuir la ansiedad y aumentar la atención.
Programas de intervención en la primera infancia, con educación especial individualizada y seguimiento en el resto del periodo educativo.
Terapia del habla y del lenguaje.
Modificación e intervención del medio, mejora y apoyo de las interacciones sociales.
Plan integral de apoyo conductual para el hogar y la escuela.
Terapias conductuales para ayudar a minimizar las conductas agresivas en el entorno escolar.
Manejo terapéutico del trastorno del sueño.
¿Cuál es la enfermedad que se da en el cromosoma 17?
Las pérdidas de material genético en zonas específicas del cromosoma 17 (parte del brazo corto del cromosoma 17, el segmento 17p11.2) dan lugar al síndrome de Smith-Magenis, llamado así por los apellidos de sus dos descubridoras.
El asesoramiento genético ofrecido a través de los resultados de los test prenatales no invasivos ayuda a diagnosticar las microdeleciones, como la producida en el síndrome de Smith-Magenis, favoreciendo el diagnóstico precoz y ofreciendo las opciones para su tratamiento y manejo oportunos.

Artículo especializado
Hematoma en la placenta: síntomas y tratamiento
Los hematomas placentarios son acúmulos de sangre que se producen por una hemorragia intrauterina en el transcurso del embarazo. Pueden dar lugar al desprendimiento de la placenta y otras complicaciones graves.
Hematomas en el embarazo
Los hematomas aparecen al desarrollarse acúmulos de sangre dentro de la cavidad endometrial, situada entre la capa más superficial del útero y el tejido corial.
No todos los hematomas intrauterinos son peligrosos. Sin embargo, la aparición de un hematoma hace que el embarazo pasa a considerarse de riesgo y se tiene una mayor precaución y control para poder valorar su evolución.
El pronóstico del hematoma depende de sus dimensiones, ubicación y el momento del diagnóstico en el embarazo (no es lo mismo un hematoma en el primer trimestre de la gestación que en el último). De este modo, un hematoma pequeño suele tener un buen pronóstico, con un escaso riesgo de aborto al inicio del embarazo. Por el contrario, un hematoma de gran tamaño ubicado en una zona peligrosa, acompañado de dolor abdominal y un sangrado abundante, en el último trimestre puede ir asociado a un riesgo elevado de desprendimiento prematuro de la placenta normoinserta o abruptio placentae . Este se define por la separación aguda parcial o completa de la placenta respecto a la pared uterina, que sucede después de la semana 20, provocando un 20 % de mortalidad fetal y hasta un 1 % de mortalidad materna.
Por otro lado, según la localización del hematoma, este se puede clasificar en hematoma subamniótico, subcoriónico y retroplacentario, de acuerdo a su relación entre la cavidad uterina y el lecho placentario .
En función de las circunstancias específicas de cada caso el médico tendrá que valorar la mejor solución.
¿Qué es un hematoma retroplacentario?
El hematoma retroplacentario (HRP) se define por el desprendimiento prematuro de la placenta normalmente insertada, de forma parcial o total.
Se trata de un sangrado de alta presión, que se produce como consecuencia de la rotura de las arterias espirales de la placenta, a nivel de la decidua basal materna o capa que recubre el útero.
El hematoma puede interrumpir en mayor o menor grado los intercambios materno-fetales y producir trastornos hemodinámicos, anomalías de la coagulación y sufrimiento fetal agudo de intensidad variable.
Se trata de una de las causas principales de hemorragia en la segunda mitad del embarazo, constituyendo una urgencia obstétrica que puede llegar a afectar hasta el 1 % de los embarazos.
En muchas ocasiones se puede sospechar la presencia de un hematoma retroplacentario debido a los signos que preceden al parto (hemorragias, anomalías del ritmo cardíaco fetal, dolor) y el diagnóstico se confirma durante el alumbramiento por la presencia de coágulos entre la cara placentaria materna y la pared uterina. Con menor frecuencia, el desprendimiento placentario es asintomático y se descubre después del parto.
La visualización ecográfica del hematoma puede servir para confirmar la sospecha diagnóstica. Sin embargo, el desprendimiento no suele tener traducción ecográfica. De igual manera, el estudio histológico de la placenta no siempre confirma el diagnóstico de desprendimiento placentario .
Causas de un hematoma retroplacentario
La formación de un HRP puede obedecer a la ruptura de una arteria decidual materna o la necrosis de una vena decidual. En todos los casos, el origen del desprendimiento de la placenta es la formación del hematoma.
Por otro lado, existen numerosos factores de riesgo de la madre para la formación de un hematoma retroplacentario, entre ellos:
Edad materna superior a 35 años o inferior a 20.
Raza negra.
Madre sola.
Tabaquismo.
Alcoholismo y consumo de drogas.
Hipertensión arterial crónica.
Hiperhomocisteinemia.
Trombofilia.
Diabetes preexistente.
Hipotiroidismo.
Anemia.
Anomalías uterinas.
Preeclampsia.
Traumatismo durante la gestación.
¿Cómo eliminar un hematoma retroplacentario?
El hematoma retroplacentario es una urgencia vital para la madre y el feto. El tratamiento dependerá de las circunstancias del diagnóstico, de la edad gestacional y del estado materno y fetal. En todos los casos, pretenderá equilibrar el estado de la madre y proceder a la evacuación del feto, en la mayoría de los casos por medio de cesárea.
Cuando se produce el desprendimiento prematuro de la placenta a término o cerca del término, el parto debe producirse con celeridad. Si el diagnóstico se sospecha antes del trabajo de parto, en general el nacimiento se debe producir por cesárea.
En cualquier caso, se trata de una urgencia obstétrica que puede requerir la finalización inmediata de la gestación. El grado de urgencia y la vía de finalización de la gestación dependerán del estado materno y fetal.
Por otro lado, cuando el hematoma retroplacentario tiene lugar en el primer trimestre suelen presentar un pronóstico favorable en más de la mitad de los casos, siendo fundamental su detección precoz.
¿Qué pasa si estoy embarazada y tengo un hematoma en la placenta?
En función de la posición y el tamaño del hematoma retroplacentario se puede producir un aborto por efecto mecánico o ciertas complicaciones gestacionales, incluyendo el desprendimiento de la placenta, un parto prematuro y el retraso en el crecimiento fetal.
Los síntomas que pueden aparecer son muy variables. Existe una forma totalmente asintomática en la que el coágulo o coágulos se descubren al examinarla placenta después del alumbramiento. Otros se manifiestan con clínica de parto pretérmino idiopático o metrorragia sin dolor. No obstante, el cuadro clínico del hematoma retroplacentario es el que acompaña al desprendimiento de placenta, con metrorragia, hipertono, la pared abdominal se hace rígida en un estado de contracción permanente y el registro cardiotocográfico fetal es patológico.
En la forma típica, la paciente siente un dolor intenso de aparición brusca. Este dolor persistente se asocia a una contractura permanente en el útero y a contracciones uterinas. La exploración en ocasiones permite visualizar pérdidas sanguíneas anómalas típicamente de aparición tardía, de poca cantidad y negruzcas. El tacto vaginal revela un cuello cervical duro.
En otros casos, existe la ausencia de sangrado uterino, presentándose como una amenaza de parto prematuro y anomalías del ritmo cardiaco fetal que se relacionan con un sufrimiento fetal agudo.
La mortalidad y la morbilidad perinatales se relacionan con la prematuridad, el CIR (crecimiento intrauterino restringido) y el sufrimiento fetal. También dependen de la extensión del desprendimiento placentario.
Artículo especializado
Inmunoglobulina: valores y análisis de sangre
Las inmunoglobulinas (Ig) o anticuerpos son sustancias del sistema inmunitario encargadas de luchar contra infecciones y enfermedades. Sus valores aumentados o disminuidos pueden indicar la presencia de una patología.
¿Qué es la inmunoglobulina y para qué sirve?
Las inmunoglobulinas son un tipo de proteínas sintetizadas por el organismo con el fin de conferir inmunidad. El sistema inmunitario fabrica anticuerpos o inmunoglobulinas para proteger al cuerpo de bacterias, virus y alérgenos.
A partir de una prueba serológica se puede proceder a la identificación en la sangre de los anticuerpos específicos encargados de neutralizar a un agente infeccioso.
Por otro lado, el desarrollo de la inmunoterapia permite que, en determinadas situaciones patológicas, se puedan administrar inmunoglobulinas por vía intravenosa para tratar determinadas enfermedades, como, por ejemplo, enfermedades oncológicas.
Las inmunoglobulinas se pueden diferenciar en 5 clases diferentes: Ig A, G, M, E y D. Las más útiles con fines diagnósticos son la IgM y la IgG:
Inmunoglobulina M (IgM). Los niveles aumentados suelen indicar infección activa por un microorganismo de forma reciente. Determinan protección a corto plazo, mientras el organismo produce otros anticuerpos. La IgM se encuentra en la sangre y en el líquido linfático.
Inmunoglobulina G (IgG). Suele indicar protección duradera contra un microorganismo. Cuando el organismo se ha visto expuesto a una infección, la recuerda con posterioridad para poder generar una respuesta inmune eficaz a partir de las IgG. Son los anticuerpos más abundantes. Cuando se vuelve a estar expuesto a un mismo microorganismo, el sistema inmunitario produce IgG específicas de forma rápida.
Cuando existe positividad de IgM e IgG de manera simultánea, suele significar que el organismo está combatiendo la infección de manera satisfactoria, luchando contra la infección activa y generando anticuerpos para generar una respuesta inmune eficaz en el futuro.
Además de las IgM e IgG, existen otras inmunoglobulinas con otras funciones específicas. Así, las inmunoglobulinas IgA están presentes en el tracto respiratorio y el sistema digestivo para proteger estos sistemas de las infecciones que les afectan. De igual modo, la IgE está presente en los pulmones, en la piel y las mucosas. En este caso, reaccionan contra sustancias identificadas como extrañas (polen, esporas de hongos, piel de animales, etc.). De este modo, la inmunoglobulina E por encima de valores normales está presente en personas con alergias.
Finalmente, por lo que respecta a la IgD, esta aparece en pequeñas cantidades en el suero humano, no de forma libre en el plasma, sino en la superficie de los linfocitos B. No es bien conocido cuál es su función específica.
¿Para qué se utiliza una prueba serológica?
Una prueba serológica de identificación de inmunoglobulinas puede tener múltiples utilidades, como valorar el funcionamiento del sistema inmune, especialmente si se padecen infecciones recurrentes.
Por otro lado, las inmunoglobulinas aumentadas pueden contribuir al diagnóstico de infecciones y patologías que pueden causar estos niveles modificados de anticuerpos, especialmente de IgM, IgG e IgA. Estas incluyen enfermedades autoinmunes, algunas neoplasias que afectan a la médula ósea o a la sangre, infecciones crónicas y algunas enfermedades congénitas.
¿Qué pasa si la inmunoglobulina está alta?
Como se ha dicho, una inmunoglobulina alta en sangre puede estar relacionada con la presencia de una infección o enfermedad.
Algunas causas de niveles elevados de una o varias inmunoglobulinas incluyen, entre otras:
Enfermedades autoinmunes (artritis reumatoide, lupus eritematoso, etc.).
Hepatitis.
Infecciones crónicas.
Ciertos tipos de cáncer.
¿Qué enfermedades se tratan con inmunoglobulina?
La terapia con inmunoglobulinas se utiliza para tratar trastornos del sistema inmune. Constituye un tratamiento eficaz para algunas enfermedades autoinmunes e inflamatorias. Normalmente, esta terapia está indicada cuando el organismo padece una inmunodeficiencia, incluyéndose diferentes situaciones que requieren que se reestablezcan los niveles de anticuerpos y se estimule el sistema inmune, como en la prevención de infecciones tras un trasplante de médula ósea.
Las inmunoglobulinas de administración intravenosa se preparan a partir de los anticuerpos presentes en la sangre de donantes sanos.
El tratamiento con inmunoglobulinas en el caso de las patologías autoinmunes funciona de manera que se evita que el propio organismo se ataque a sí mismo y para disminuir la inflamación.
Su prescripción está contemplada en diversos tipos de patologías:
Inmunodeficiencias primarias. El tratamiento sustitutivo de los síndromes de inmunodeficiencia primaria con escasa o ninguna producción de inmunoglobulinas. En estos casos, los anticuerpos aportados consiguen una prevención eficaz frente a infecciones bacterianas, cuya incidencia disminuye notablemente tras el tratamiento.
Inmunodeficiencias secundarias. Incluyen diversos estados de inmunodeficiencia humoral secundaria, tales como neoplasias y SIDA.
Otras indicaciones. El uso de inmunoglobulinas intravenosas, debido a sus propiedades inmunomoduladoras y antiinflamatorias se aplica también en enfermedades hematológicas, infecciosas, trastornos neuroinmunológicos y autoinmunes. De este modo, las inmunoglobulinas intravenosas se utilizan para tratar ciertas enfermedades reumáticas. También se puede utilizar en el tratamiento de enfermedades musculares inflamatorias y puede usarse para tratar recuentos plaquetarios muy bajos en pacientes con lupus eritematoso sistémico. Además, puede resultar útil en el tratamiento de muchas otras afecciones por inmunodeficiencia.
Artículo especializado
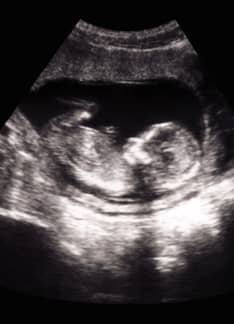
Ecografía de las 20 semanas: ¿Qué detecta?
La ecografía es una técnica por imagen no invasiva que permite hacer un seguimiento adecuado del desarrollo fetal durante el embarazo. A lo largo de todo el periodo de gestación, es recomendable llevar a cabo al menos tres exploraciones ecográficas.
Una de ellas es la exploración ecográfica del segundo trimestre o ecografía de las 20 semanas. Se trata de una prueba fundamental que permite confirmar el desarrollo normal del bebé y es útil para identificar posibles malformaciones.
La vía abdominal es la preferente y la utilizada clásicamente para la ecografía de las 20 semanas.
¿Que se ve en la eco de las 20 semanas?
La ecografía de las 20 semanas o ecografía morfológica se realiza en todas las embarazadas en el segundo trimestre de embarazo, alrededor de la semana 20 (de la 18 a la 22). A esta edad gestacional ya se ha producido un desarrollo suficiente de los órganos y tejidos fetales, por lo que es posible detectar anomalías mayores.
La ecografía se realiza por medio de un ecógrafo, cuya sonda de ultrasonidos se sitúa sobre el abdomen de la madre. El eco de los ultrasonidos, tras penetrar en los tejidos de diferente densidad, se transforman en imágenes, pudiendo observar los órganos y estructuras fetales para valorar su estado.
De este modo, se obtiene la máxima información sobre el desarrollo fetal, identificando posibles malformaciones.
La prueba suele estar protocolizada e incluye una serie de mediciones y estudios anatómico-morfológicos:
Confirmación del latido cardíaco del feto, incluyendo la visualización del corazón y su estructura anatómica.
Valoración de la cantidad de líquido amniótico y la placenta.
Estudio pormenorizado de la anatomía fetal. Este incluye diferentes biometrías cefálicas (tamaño del cráneo y diferentes partes de la cabeza). También se estudia el cuello, la columna, el tórax (pulmones y diafragma), abdomen (estómago, vesícula biliar, intestinos, riñones y vejiga) y las extremidades para descartar cualquier malformación o alteración morfológica.
Evaluación del crecimiento fetal, teniendo en cuenta la edad gestacional estimada en el primer trimestre.
Análisis de la estructura uterina y las zonas anatómicas próximas.
Estudio de los genitales, por lo que se puede averiguar el sexo del bebé.
La duración del estudio es de unos 20-30 minutos en su totalidad.
¿Cuál es la ecografía más importante en el embarazo?
Durante toda la gestación se recomienda llevar a cabo al menos 3 estudios ecográficos. Todos ellos tienen su relevancia y razón de ser.
La primera de ellas, en torno a la semana 12 de gestación, evalúa, principalmente, el riesgo de cromosomopatías y síndromes genéticos. En la tercera, entre las semanas 34 y 36 del embarazo, se evalúa, principalmente el crecimiento y desarrollo fetal.
Finalmente, la ecografía de la semana 20 es señalada por la Organización Mundial de la Salud como necesaria para calcular la edad gestacional y mejorar la detección de anomalías fetales y embarazos múltiples. Además, esta permite a los padres una mayor capacidad de decisión en relación con las diferentes opciones planteadas a partir de la eventual detección de alguna anomalía.
¿Cómo está un bebé en la semana 20?
En la semana 20 de gestación el feto ya es lo suficientemente grande como para poder observar sus órganos con claridad. El grado de desarrollo de los órganos y tejidos permite la detección de anomalías mayores.
Órganos como el corazón, los riñones, el hígado, el intestino, los genitales o la columna vertebral ya tienen un grado de desarrollo importante. Tanto la forma como sus proporciones son parecidas a las que presentará en el momento del nacimiento.
En este momento de la gestación ya se pueden estudiar las cavidades del corazón (aurículas y ventrículos), además de la estructura de los riñones, el hígado, el estómago y otros órganos. También se pueden contar las vértebras de la columna y los dedos de manos y pies (si falta alguno puede ser el signo de otros problemas más graves).
Además, se puede analizar que no existan fisuras en el paladar, la presencia de cristalino en los ojos y la existencia de los huesos largos en las extremidades, así como verificar la alineación de los huesos largos y extremidades inferiores con los pies.
En la epidermis, la capa más superficial de la piel, se empiezan a formar los surcos característicos determinados genéticamente de las manos y plantas de los pies.
Por otro lado, en este momento se puede calcular la circunferencia del abdomen y la cabeza para valorar el crecimiento fetal. Con esta y otras mediciones se puede dirimir el tiempo exacto de gestación, junto con la fecha probable de parto.
Ecografía 3D, 4D y 5D
La ecografía obstétrica utilizada para el seguimiento y diagnóstico de los diferentes problemas de desarrollo del feto está basada en una tecnología “2D”, es decir, imágenes en blanco y negro que ofrecen secciones de los tejidos y órganos. Esta técnica permite explorar el feto para analizar su anatomía e identificar malformaciones.
Además de esta tecnología originaria 2D, hace algunos años se desarrolló la tecnología 3D, permitiendo a partir del procesamiento informático de un barrido en dos dimensiones sobre un tejido concreto, formar una visualización de tres dimensiones. A partir de aquí, se pudo avanzar para ver estas imágenes 3D en movimiento en tiempo real, dando lugar a la aparición de la ecografía 4D.
De hecho, se puede realizar una ecografía de las 20 semanas 4D, ya que los aparatos de alta gama empleados para la ecografía morfológica incorporan diferentes programas 3D y 4D para complementar el estudio de las diferentes estructuras de órganos y tejidos.
Si bien el modo 2D es la base de la ecografía del segundo trimestre, en algunos casos, la aplicación de estas nuevas tecnologías puede resultar de utilidad y de especial interés para el estudio del sistema nervioso, corazón, esqueleto y otros tejidos y sistemas.
Finalmente, la ecografía 5D es otra evolución en la que la mejora del tratamiento de la imagen le otorga mayor variedad de tonos y textura, dando a la imagen un aspecto más real. De este modo, la ecografía 5D de las 20 semanas ofrece imágenes más vistosas, pero sin aportar mejores elementos diagnósticos.

Artículo especializado
Mosaicismo genético: ¿Cómo detectarlo?
Frente a las aneuploidías o anormalidades cromosómicas por exceso o defecto de cromosomas que pueden determinar la aparición de diferentes síndromes congénitos, el mosaicismo genético se da cuando el embrión posee una mezcla heterogénea de células cromosómicamente normales y anormales.
¿Qué es el mosaicismo y cómo se produce?
El mosaicismo cromosómico o mosaicismo fetal es una alteración genética en la que un individuo presenta 2 o más grupos de células con distinta composición genética, supuestamente originadas a partir de un mismo cigoto. Esta afección puede alterar cualquier tipo de célula, incluyendo células sanguíneas, óvulos, espermatozoides y células de la piel.
Cromosomas
La información genética presente en el ADN se organiza dentro de cada célula en los cromosomas. Los seres humanos tenemos 23 pares de cromosomas (dos copias de cada par, es decir 46). Los primeros 22 pares se numeran del 1 al 22. El último par determina el sexo. Las niñas tienen dos cromosomas X y los niños un cromosoma X y un cromosoma Y. Los problemas de salud y de desarrollo aparecen cuando falta o sobra algún cromosoma o una parte de ellos. En el caso del mosaicismo, este exceso o defecto cromosómico no se da en todas las células.
Causas del mosaicismo
Las causas del mosaicismo suelen estar relacionadas con accidentes acaecidos durante la división celular del cigoto durante la gestación, en fases muy tempranas del desarrollo fetal y no parece estar asociada a ningún factor paterno ni materno. Las causas pueden ser de 2 tipos:
En la fase de la división celular en la que los pares de cromosomas son separados (anafase), un cromosoma podría retrasarse durante el momento del replicado cromosómico. De este modo, este cromosoma quedaría excluido, dando como resultado después de la división una célula con un cromosoma normal y otra célula con un cromosoma de menos. Si la pérdida de dicho cromosoma no interfiere con la viabilidad y replicación de la célula, ambas seguirán replicándose y el nuevo individuo presentará un mosaicismo de ambos tipos celulares (normales y anormales).
En un segundo tipo de accidente, un cromosoma se replicaría sin dividirse adecuadamente en dos partes en movimiento hacia los polos opuestos de la célula, yendo las dos mitades al mismo polo. El resultado sería tras la división una célula con un cromosoma extra y una célula con un cromosoma de menos.
De este modo, el mosaicismo afecta a la información genética de unas células, pero no de todas. Un individuo que presenta una trisomía en mosaico significa que algunas de sus células tienen tres copias de un cromosoma, mientras que otras tienen dos, es decir, son normales.
Por ejemplo, el mosaicismo cutáneo se daría cuando la afectación de las células de la piel determinara dos o más tipos de células genéticamente distintas que darían diferentes tipos de piel mezcladas. Estos tipos de células producirían diferente cantidad de pigmento, resultando en áreas de piel con distintas tonalidades, por la distinta cantidad de melanina genéticamente programada en las diferentes líneas celulares.
Mosaicismo de línea germinal
El mosaicismo de línea germinal hace referencia al mosaicismo que afecta a los óvulos y al esperma. De este modo, algunos óvulos o espermatozoides tienen una configuración cromosómica normal y otros no. Si uno de los óvulos o espermatozoides afectados participara en un proceso fecundativo, el niño podría presentar una anomalía cromosómica.
No es posible determinar si una persona tiene mosaicismo de línea germinal.
Mosaicismo placentario
El mosaicismo placentario o mosaicismo confinado a la placenta se define como la presencia de dos o más líneas celulares genéticamente diferentes que están circunscritas a la placenta y no están presentes en el feto.
¿Cómo identificar un mosaicismo?
El mosaicismo genético se puede diagnosticar de diferentes maneras. Cuando existe mosaicismo en un feto que se está gestando, puede haber células del feto con diferente composición cromosómica en la sangre de la madre. En este caso, los test prenatales no invasivos a partir de una muestra de sangre materna pueden ser problemáticos y dar como resultado un falso negativo.
En el caso de análisis de sangre fetal, el diagnóstico puede ser más complicado en función de las células afectadas por el cambio cromosómico. Por ejemplo, un análisis de sangre puede servir para diagnosticar mosaicismo si el cambio cromosómico está ubicado en células como las de la piel.
¿Qué diferencia hay entre el síndrome de Down y el mosaico?
En general, las personas con mosaicismo presentan los mismos problemas que las que presentan un síndrome por anomalías cromosómicas sin mosaicismo. De este modo, una persona con trisomía 21p en mosaico posiblemente tenga muchas de las mismas características que las personas con trisomía 21p sin mosaico (síndrome de Down). Sin embargo, debido a que los individuos con mosaicismo tienen menos células con la anomalía genética, muchos de los síntomas asociados al síndrome pueden aparecer de una forma más leve.
Desafortunadamente, no es posible predecir con exactitud de qué modo el mosaicismo puede llegar a afectar a una persona, ya que no se puede saber con exactitud las células que presentan la anomalía genética.

Artículo especializado
Síndrome de Klinefelter: ¿Qué es y cómo detectarlo?
Las aneuploidías son anomalías que aparecen cuando se produce un exceso o defecto en los cromosomas. Las que afectan a los cromosomas sexuales son las más habituales. Entre ellas destaca el síndrome Klinefelter, un trastorno genético en el que aparece un cromosoma X adicional en los hombres. Se trata de un trastorno congénito, pero en ocasiones no se diagnostica hasta la edad adulta.
¿Qué produce el síndrome de Klinefelter?
El síndrome de Klinefelter es la primera anomalía de los cromosomas sexuales que ha sido descrita en humanos y que tiene una incidencia de 1 de cada 1.000 varones nacidos.
Cada célula del cuerpo humano contiene en su información genética 23 pares de cromosomas, es decir, 46. La mitad proviene de la madre y la otra mitad del padre. Los cromosomas contienen el material genético con la información necesaria para el correcto desarrollo y funcionamiento del organismo. Los pares de cromosomas del 1 al 22 se denominan autosomas (ADN autosómico) y el par 23 (XX en las mujeres y XY en los hombres) constituyen los cromosomas sexuales.
El síndrome de Klinefelter no se hereda, sino que ocurre como un evento aleatorio durante la formación de las células reproductivas ( óvulos y espermatozoides) que tiene como resultado la presencia de una copia extra del cromosoma x en cada célula (47,XXY).
El síndrome de Klinefelter solo tiene lugar en varones. En un 75 % de los casos se produce un cariotipo XXY. Aproximadamente en un 20 % se alternan diferentes composiciones cromosómicas (mosaicos cromosómicos), es decir, XY en el cromosoma sexual en unas células y XXY en otras. También existen otras variantes minoritarias que incluyen las combinaciones XXYY, XXXY, y XXXXY en los cromosomas sexuales.
Las alteraciones de los cromosomas sexuales suelen ser fenómenos aislados, en los que el único factor predisponente conocido es la edad materna avanzada.
¿Cómo son los niños con síndrome de Klinefelter?
Las aneuploidías de los cromosomas sexuales se caracterizan por una gran variabilidad clínica entre individuos. En cualquier caso, suele tratarse de individuos altos y delgados, con piernas largas. Físicamente no aparecen signos diferenciales hasta llegar a la pubertad. En ese momento empiezan a observarse signos de hipogonadismo, que impide que el cuerpo produzca esperma y testosterona, con tendencia a la obesidad. Esto significa que con frecuencia en la edad adulta serán estériles en un 70 % los casos.
Las deficiencias y anormalidades se hacen más notables cuando aparecen más de dos cromosomas X. En estos casos el desarrollo sexual es más deficiente y el déficit intelectual más grave.
Los signos distintivos más frecuentes en preadolescentes afectados son unos genitales pequeños y unas piernas largas.
En bebés y niños de corta edad en ocasiones no se aprecian signos o estos son muy leves e inespecíficos. Pueden presentar músculos débiles con un desarrollo motor lento y retraso en el habla.
¿Cómo se puede identificar el síndrome de Klinefelter?
El médico puede sospechar la presencia del síndrome de Klinefelter cuando se dan los siguientes síntomas y signos:
Niño con retraso leve y comportamiento inmaduro. La mayoría presentan problemas de aprendizaje, especialmente dislexia. El lenguaje y la memoria auditiva son deficientes. Además, presentan dificultades para leer y escribir.
Los trastornos del comportamiento son frecuentes, especialmente inmadurez, inseguridad y timidez. Las personas afectadas suelen presentar problemas para relacionarse con individuos de su grupo de edad y de adaptación social. La depresión es frecuente en estos individuos.
Adolescente con testículos pequeños. Los caracteres sexuales secundarios se desarrollan poco. El vello corporal es escaso y la distribución puede ser ginecoide, es decir que se desarrolla en zonas típicas femeninas. El tejido graso subcutáneo también puede adoptar una distribución femenina sobre todo a nivel de las caderas, y pueden presentar ginecomastia o agrandamiento de las mamas.
Adulto con hábito eunucoide (órganos sexuales subdesarrollados con afectación en la producción hormonal testicular), ginecomastia y escaso desarrollo muscular. Debido a esta masa muscular poco desarrollada, el cansancio es un síntoma habitual.
Adulto con infertilidad.
Son comunes la aparición de varices y úlceras en las extremidades inferiores.
Existe un mayor riesgo de desarrollar enfermedades autoinmunes.
Los varones con cromosomas sexuales XXY con ginecomastia tienen mayor riesgo de cáncer de mama.
Si bien muchos de los casos de síndrome de Klinefelter no se identifican hasta la edad adulta, con el desarrollo de las técnicas de análisis genético, en los últimos años muchos casos se diagnostican en la etapa prenatal. De este modo, existen test prenatales no invasivos que pueden detectar las anomalías cromosómicas, incluyendo las relacionadas con los cromosomas sexuales como el síndrome de Klinefelter, analizando el ADN fetal presente a partir de una muestra sanguínea materna.
Tratamiento del síndrome de Klinefelter
No existe un tratamiento que pueda revertir las anomalías cromosómicas como las presentes en le síndrome de Klinefelter. Sin embargo, las personas afectadas pueden desarrollarse y llevar una vida normal. Para ello, será necesaria una intervención precoz que contemple un ambiente positivo en casa y en la escuela con métodos de estudio adaptados. También debe incluir un apoyo cognitivo y psicológico adecuado, junto con un seguimiento médico que incluya el tratamiento hormonal.
La terapia hormonal debe incluir el tratamiento sustitutivo con testosterona desde que se inicia la pubertad. Este será necesario para promover el desarrollo de los caracteres sexuales secundarios masculinos, el crecimiento testicular y el aumento de la masa muscular.
En cuanto a la esterilidad, no existe ningún tratamiento que pueda revertirla, si bien se puede potenciar la fertilidad cuando existe una producción mínima de espermatozoides.
Artículo especializado
Aneuploidías: ¿Qué son y cómo se detectan?
Las aneuploidías son anomalías cromosómicas que pueden determinar la aparición de diferentes síndromes congénitos. Los más habituales se pueden detectar mediante la realización de un test prenatal no invasivo.
¿Qué es aneuploidía y ejemplos?
Las aneuploidías son las alteraciones genéticas en las que el número de cromosomas no son los habituales, tanto por exceso como por defecto. De este modo, existen uno o más cromosomas adicionales o faltan uno o más cromosomas.
Cada célula del cuerpo humano contiene en su información genética 23 pares de cromosomas, es decir, 46 cromosomas. La mitad proviene de la madre y la otra mitad del padre. Los cromosomas contienen el material genético con la información necesaria para el correcto desarrollo y funcionamiento del organismo. Los pares de cromosomas del 1 al 22 se denominan autosomas (ADN autosómico) y el par 23 (XX en las mujeres y XY en los hombres) constituyen los cromosomas sexuales.
Las aneuploidías más comunes son las trisomías. En los nacidos vivos, las aneuploidías más frecuentes son el síndrome de Down (trisomía del cromosoma 21) y el síndrome de Klinefelter (trisomía del cromosoma sexual XXY).
¿Qué provoca las aneuploidías?
La mayoría de estas alteraciones cromosómicas, incluyendo las aneuploidías, se producen durante la producción de los espermatozoides y óvulos. Si un óvulo o espermatozoide defectuoso participa en la fecundación, el embrión se verá afectado por la anomalía. Sin embargo, en la mayoría de los casos se producirá un aborto espontáneo, ya que la mayoría de las aneuploidías son incompatibles con la vida.
¿Cuáles son los tipos de aneuploidías?
Desde un punto de vista teórico, las aneuploidías podrían incluir:
Nulisomías. Cuando faltan los 2 cromosomas homólogos.
Monosomías. Cuando falta un cromosoma.
Disomías. Cuando el número de cromosomas es correcto, pero un par de cromosomas provienen del mismo progenitor.
Trisomías. Cuando en un par de cromosomas se une un tercero.
Tetrasomías y pentasomías. Existen 2 o 3 cromosomas extra, siempre en los cromosomas sexuales.
Las aneuploidías más frecuentes en humanos son las trisomías y monosomías:
Trisomías. En los nacidos vivos las aneuploidías más frecuentes son el síndrome de Down (trisomía del cromosoma 21), seguido por el síndrome de Klinefelter (trisomía XXY).
Monosomías. El más habitual es el síndrome de Turner (monosomía X). No son compatibles con la vida las monosomías de los cromosomas autosómicos.
El resto de las trisomías más comunes, como el síndrome de Edwards (trisomía 18) o el síndrome de Patau (trisomía 13), son incompatibles con la vida y presentan múltiples malformaciones detectables por ecografía. De este modo, el síndrome de Down es la aneuploidía más común causante de discapacidad en la que se enfoca el cribado durante el embarazo.
Aneuploidías autosómicas
Las aneuploidías autosómicas compatibles con la vida dan lugar a síndromes genéticos con importantes consecuencias en el desarrollo del bebé. Se incluyen aquí:
Trisomía 21 (síndrome de Down). Se da en aproximadamente 1 de cada 700 nacimientos. El riesgo aumenta con la edad de la madre. Suele provocar retraso mental y también malformaciones físicas, especialmente defectos cardíacos.
Las trisomías 18 (síndrome de Edwards) y 13 (síndrome de Patau) son más raras y se encuentran en aproximadamente 1 de cada 7.000 nacimientos. El riesgo también se ve aumentado con la edad de la madre. Estas trisomías se asocian a un grave retraso mental y a menudo a graves malformaciones físicas. En la mayoría de los casos no sobreviven al primer año de vida.
Aneuploidías de los cromosomas sexuales
Las aneuploidías de los cromosomas sexuales consisten en la presencia de alteraciones que pueden tener distintas consecuencias, incluyendo malformaciones, trastornos cognitivos y de crecimiento, así como problemas sexuales y de infertilidad.
Síndrome de Klinefelter. Afecta a los hombres y está causado por la presencia de un cromosoma X adicional (XXY). El riesgo aumenta con la edad de la madre. Los afectados suelen presentar una estatura elevada, un ligero retraso mental y problemas de fertilidad.
Síndrome de Jakob. Afecta a los hombres y está causado por la presencia de un cromosoma Y adicional (XYY). Los afectados también presentan una altura mayor de lo normal y pueden presentar retraso en el lenguaje.
Síndrome de Turner. Afecta a las mujeres y está definido por la ausencia de un cromosoma X (X0). Esta monosomía determina baja estatura, infertilidad y trastornos cardiacos y renales.
Síndrome triple X. Afecta a las mujeres y se caracteriza por la presencia de un cromosoma X de más (XXX). En muchas ocasiones no se manifiestan síntomas o estos son leves. Estas mujeres suelen tener una fertilidad normal salvo que se vean afectadas por fallo ovárico precoz o presenten malformaciones genitourinarias que impidan la gestación.
Todavía no ha sido correctamente estudiado si los pacientes tienen más riesgo de que su descendencia, en el caso de que esta sea posible, también presente trisomía (especialmente, triple X o Klinefelter).
Microdeleciones
Las microdeleciones son pequeñas pérdidas de material genético que se asocian con graves problemas del desarrollo en el recién nacido. Incluyen el síndrome de DiGeorge, el síndrome de deleción 1p36, el síndrome de Cri-du-chat, el síndrome de Angelman y el síndrome de Prader-Willi.
El síndrome por microdeleción más frecuente es el de DiGeorge. En él está ausente un pequeño fragmento del cromosoma 22 (22q11.2), lo que acarrea múltiples alteraciones fetales como defectos cardíacos, anomalías en el paladar y la cara, retraso en el desarrollo e inmunodeficiencia, con infecciones frecuentes.
Triploidía
La triploidía es una alteración cromosómica en la que el feto tiene tres copias de cada cromosoma en lugar de dos (69 cromosomas y no 46). Normalmente se produce cuando dos espermatozoides fertilizan un óvulo, pero también puede ocurrir cuando un óvulo diploide es fertilizado por un espermatozoide, o bien en casos en los que un espermatozoide diploide fertiliza un óvulo. Las triploidías no son compatibles con la vida.
Detectar las aneuploidías mediante test prenatales no invasivos
Para el diagnóstico prenatal de las anomalías genéticas más comunes se requiere la obtención de material genético del feto mediante una técnica invasiva como la amniocentesis o la biopsia corial. Se trata de pruebas que llevan asociado un riesgo de pérdida gestacional, por lo que no se recomiendan de forma indiscriminada a todas las mujeres embarazadas. Este tipo de pruebas invasivas están indicadas ante un resultado de alto riesgo en un test prenatal no invasivo o en el cribado combinado de primer trimestre que se realiza de forma generalizada en la semana 12 de gestación.
Sin embargo, gracias a que el ADN del feto circula por el torrente sanguíneo de la madre durante el embarazo y el avance de técnicas genéticas, a partir de una muestra de sangre materna se puede analizar el ADN libre del feto para detectar posibles anomalías cromosómicas.
Los test prenatales no invasivos permiten comprobar el estado del bebé antes de su nacimiento e identificar posibles aneuploidías, así como conocer el sexo fetal.

Artículo especializado
Síndrome DiGeorge: ¿Qué es y cómo detectarlo?
Las anomalías cromosómicas pueden dar lugar a diversos síndromes y problemas de salud. En el caso de la trisomía del cromosoma 21 (presencia de 3 cromosomas en lugar de los 2 preceptivos) el bebé nace con síndrome de Down. De igual modo, la falta de un cromosoma o una parte de él, también se producen complicaciones de salud de origen genético.
Es el caso del síndrome DiGeorge, también conocido como síndrome de deleción del cromosoma 22q11.2, esta pérdida provoca el desarrollo insuficiente de varios sistemas del organismo. Este síndrome abarca varios términos que antes se consideraban afecciones separadas, como el síndrome de Di George, el síndrome velocardiofacial o el síndrome cardiofacial.
¿Qué produce el síndrome de DiGeorge?
En condiciones normales existen dos copias de los 23 cromosomas, cada una heredada de un progenitor. Cuando a una de las copias del cromosoma 22 le falta el segmento 22q11.2 (30-40 genes) esto da lugar a la aparición del síndrome de DiGeorge de manera innata (anomalía del brazo corto del cromosoma 22).
La eliminación de estos genes del cromosoma 22 se produce de forma aleatoria en el espermatozoide o el óvulo de los progenitores, o puede tener lugar en las primeras etapas de la gestación.
La deleción o ausencia de esta parte del cromosoma da lugar a la aparición de trastornos y anormalidades en diferentes órganos y sistemas del organismo.
Se ha calculado que en un 10% de las familias con un caso de síndrome de DiGeorge es por causa hereditaria y se comporta de manera autonómica dominante, es decir, que si existen antecedentes familiares, hay un 50% de posibilidades de transmitirla a los hijos.
¿A qué órganos afecta el síndrome de DiGeorge?
Los defectos congénitos cardíacos y las anomalías en el paladar están presentes en más de un 70% de los casos.
De este modo, los problemas de salud que se asocian al síndrome de DiGeorge pueden ser variables e incluyen anomalías en diferentes órganos y sistemas, incluyendo entre otros:
El corazón.
El sistema inmunitario.
El sistema hormonal.
Aparición de deformidades en el paladar.
Bajos niveles de calcio en la sangre que dan lugar a diferentes complicaciones.
Retrasos en el desarrollo.
Problemas de aprendizaje, emocionales y trastornos de conducta.
Problemas cardíacos
Los defectos cardíacos que provoca el síndrome de deleción del cromosoma 22q11.2 podrían determinar un suministro de oxígeno insuficiente en los tejidos. Este puede venir determinado por diferentes anomalías en la estructura del corazón y de sus vasos sanguíneos. Es el caso de la tetralogía de Fallot, presente en el 92% de los casos.
Trastornos del sistema inmunitario
El timo es la glándula donde maduran las células T, un tipo de leucocitos necesarios para combatir las infecciones. El síndrome de deleción del cromosoma 22q11.2 determina una disfunción del timo que impide que el sistema inmunitario pueda luchar contras las infecciones con normalidad. Además de las infecciones recurrentes durante la infancia, el riesgo de padecer enfermedades autoinmunes durante la edad adulta también se ve aumentado.
Trastornos hormonales
El síndrome de DiGeorge puede determinar un funcionamiento reducido de las glándulas paratiroideas, lo que provoca hipoparatiroidismo. Este trastorno hace que estén presentes niveles reducidos de calcio y niveles elevados de fósforo en la sangre. Esto puede desembocar en dolores musculares, problemas renales, arritmias cardiacas y problemas en el desarrollo físico y mental, entre otras consecuencias.
Anomalías en el paladar y la cara
En el síndrome de DiGeorge también se suelen ver afectados el paladar y la cara, con la aparición de una hendidura en el paladar y la aparición de labio leporino. En estos, el tejido del paladar y del labio superior presentan una deficiencia y no quedan perfectamente unidos, lo que puede ocasionar dificultades en la deglución y en el habla, entre otros problemas.
Además, pueden aparecer ciertos rasgos faciales distintivos. Estos pueden incluir orejas pequeñas de implantación baja, abertura de los ojos reducida, rostro alargado y estrecho y nariz tubular con punta bulbosa, entre otros.
Problemas de desarrollo cerebral y aprendizaje
La deleción del cromosoma 22q11.2 puede causar problemas en el desarrollo mental y el aprendizaje, la conducta y el comportamiento social. Además, existe un mayor riesgo de trastorno por déficit de atención e hiperactividad (TDAH) y trastornos del espectro autista en la etapa infantil. En la edad adulta existe un mayor riesgo de depresión, ansiedad y otros trastornos mentales.
¿Cómo se detecta el síndrome DiGeorge?
Para el diagnóstico prenatal de las anomalías genéticas más comunes, incluyendo la deleción del cromosoma 22q11.2, se requiere la obtención de material genético del feto mediante una técnica invasiva como la amniocentesis o la biopsia corial. Estas pruebas llevan asociado un riesgo de pérdida gestacional, por lo que no se utilizan de forma indiscriminada en todos los embarazos.
En este sentido, el avance en las técnicas genéticas ha permitido desarrollar test prenatales no invasivos que se pueden aplicar como pruebas de cribado a partir de la sangre de la madre. Con ellos es posible identificar los embarazos con un riesgo elevado de presentar una anomalía genética, pero sin poner en riesgo al feto ni a la madre.
Con todas las pruebas que se realizan durante el embarazo , incluyendo las ecografías y controles periódico, además del acceso a test prenatales no invasivos, es muy complicado que una anomalía cromosómica como el síndrome de DiGeorge pase desapercibida.
¿Cuál es la función del cromosoma 22?
Los genes que forman parte del cromosoma 22 están implicados en múltiples funciones y codifican para multitud de órganos y tejidos del organismo. Por lo tanto, sus anomalías y deficiencias pueden tener consecuencias muy diversas, provocando patologías y afecciones en múltiples sistemas del organismo, como las alteraciones cardíacas, el déficit inmunitario y las características dismórficas faciales anteriormente descriptas.

Artículo especializado
Apendicitis en niños: Síntomas y tratamiento
La apendicitis es un proceso inflamatorio agudo que afecta al apéndice. Es la primera urgencia quirúrgica y una causa frecuente de dolor abdominal agudo en los niños.
El apéndice vermiforme es un pequeño trozo de tejido de tamaño variable conectado a la parte inicial del colon. Está situado en la zona inferior derecha del abdomen y desempeña un papel fisiológico como reservorio de la flora intestinal (microbiota intestinal), y en él se originan células madre mesenquimales del colon.
Los casos de apendicitis aguda varían mucho en su presentación clínica y el diagnóstico se hace más difícil por la multiplicidad de diagnósticos diferenciales en los niños. Puede iniciarse con un cuadro clínico que es común a muchos procesos abdominales, incluyendo entre otros:
Intususcepción.
Divertículo de Meckel.
Gastroenteritis víricas y bacterianas.
Hasta la mitad de los casos en niños se presentan con síntomas inespecíficos. De este modo antes de un diagnóstico definitivo debe mantenerse al paciente en observación por un tiempo prudencial.
Suele ser más común en la etapa infantil y durante la primera etapa adulta (entre los diez y los 30 años de edad). El pico de incidencia se produce entre la escuela primaria y la adolescencia, de los 10 a 19 años.
¿Cómo se produce la apendicitis?
La causa de la obstrucción del apéndice puede ser debida a la inflamación de la pared, la presencia de fecalito (heces duras) y apendicolitos (depósitos calcificados). Otro motivo puede ser la hiperplasia linfoide. En esta, el abundante tejido linfoide del apéndice en la mucosa y la submucosa determina la propensión a desarrollar una hiperplasia que provoca una obstrucción del lumen del apéndice.
La obstrucción puede provocar un aumento de la presión en el apéndice. Este continúa segregando líquido de la mucosa, lo que provoca la distensión e inflamación de los tejidos. La alterada vascularización del órgano, el sobrecrecimiento bacteriano y la eventual perforación son consecuencias de la distensión. Es un proceso progresivo en el que los síntomas del niño o adolescente empeoran hasta que se produce la perforación con el riesgo de peritonitis, es decir, que se produzca la difusión de la infección por todo el abdomen. Por tal motivo, la aparición de una apendicitis requiere de atención médica de manera urgente.
Síntomas de apendicitis en niños
El síntoma más común de la apendicitis es el dolor abdominal agudo. Se trata de un dolor alrededor del ombligo que suele aparecer después de síntomas inespecíficos como febrícula. El dolor migra entonces al cuadrante inferior derecho del abdomen, puede aumentar al caminar o al hacer movimientos bruscos.
También pueden presentarse náuseas o vómitos después de la aparición del dolor. Y también síntomas urinarios y perdida del apetito.
Los antecedentes habituales son síntomas atípicos o vagos, como dolor abdominal difuso asociado a fiebre baja. La presencia de diarrea puede retrasar el diagnóstico si los síntomas se confunden con una gastroenteritis.
Diagnóstico de la apendicitis
En primer lugar, el médico puede sospechar de la presencia de apendicitis en función de los síntomas que presente el niño.
Un cambio en la localización del dolor desde el abdomen superior al cuadrante inferior derecho se asocia a menudo con la apendicitis. En los niños y adolescentes, el interrogatorio y la exploración física deben adaptarse a la edad y a la etapa de desarrollo del paciente. La experiencia del examinador es importante, especialmente cuando el paciente es un niño pequeño. En los niños, la ausencia de náuseas y vómitos, sensibilidad abdominal y leucocitosis descarta la apendicitis con una fiabilidad del 98%.
La clave para diagnosticar una apendicitis es la palpación del abdomen. Existen ciertos puntos y maniobras de palpación que ayudan al médico a diagnosticar apendicitis (punto de McBurney’s, signo de Blumberg, signo de Rovsing y del psoas).
También se pueden realizar otras pruebas complementarias para apoyar o confirmar el diagnóstico:
En un análisis de sangre es frecuente que aparezcan unos niveles aumentados de leucocitos con neutrofilia y marcadores elevados de inflamación como la proteína C reactiva.
Técnicas de imagen como una ecografía abdominal, una radiografía, una tomografía computarizada o una resonancia magnética del abdomen pueden ser de utilidad para confirmar la apendicitis. La ecografía es el método de primera elección, especialmente en los niños, aunque la tomografía es superior para diagnosticar apendicitis y sus complicaciones, principalmente cuando hay dudas con el diagnóstico.
¿Cómo se trata la apendicitis?
El tratamiento de la apendicitis suele ser quirúrgico y consiste en la extirpación del apéndice (apendicectomía). Cuando es posible su aplicación, se pueden utilizar técnicas quirúrgicas por laparoscopia, menos invasivas que la cirugía abierta. En los niños, también se ha descrito una técnica mínimamente invasiva para la apendicetomía asistida por laparoscopia transumbilical. La apendicetomía laparoscópica asistida por un solo puerto, aunque todavía no se ha convertido en el estándar de oro, también se ha realizado en niños y se ha comprobado que es segura.
Recientemente se ha planteado el tratamiento conservador con antibióticos en apendicitis no complicadas, sin embargo, esta práctica no está todavía bien reconocida ni establecida.
Ante la sospecha de apendicitis, el paciente no debe recibir nada por la boca y se indican antibióticos de manera profiláctica.
En ocasiones, las pruebas realizadas no aciertan con el diagnóstico, por lo que no es excepcional que en el transcurso de la operación se compruebe que el apéndice no está afectado.
En una apendicitis no complicada la operación quirúrgica suele ser sencilla y el tiempo de recuperación es relativamente corto. Si el apéndice se ha visto perforado, la operación es más compleja y existe más riesgo de complicaciones, por lo que la recuperación es más lenta y precisa de más días de ingreso hospitalario. La estancia posoperatoria varía, según la gravedad del cuadro. Las complicaciones pueden incluir los abscesos de la pared abdominal, hemorragias, obstrucción intestinal, absceso intrabdominal, etc. De ahí la importancia de realizar un diagnóstico y tratamiento adecuado en las primeras 24-48 horas tras el inicio de los síntomas.
Artículo especializado
Monocitos altos: ¿Qué niveles son preocupantes?
Los glóbulos blancos o leucocitos son un tipo de células sanguíneas cuya función es defender al organismo frente a infecciones, agentes patógenos y células tumorales.
Existen varios tipos de leucocitos: linfocitos, eosinófilos, neutrófilos, basófilos y monocitos .
Los valores de glóbulos blancos se pueden expresar en el hemograma (o análisis de sangre) de dos modos: En valor absoluto, es decir, la cifra del total de células que se encuentran en la muestra, o en valor relativo al resto de tipos de linfocitos.
Tienen una morfología muy concreta, aunque en ocasiones se pueden confundir con otros leucocitos .
¿Qué son los monocitos altos?
Los monocitos son un tipo de leucocitos que son producidos por la médula ósea, desde donde pasan al torrente sanguíneo. Cuando llegan a un tejido específico se transforman en macrófagos, encargados de aislar y eliminar microorganismos. También eliminan células muertas, restos celulares, junto con sustancias extrañas. Se trata de un componente fundamental de la respuesta inmune.
De este modo, los macrófagos se pueden definir como “células limpiadoras” del sistema inmunitario.
Su actuación está mediada a través de dos funciones:
La fagocitosis, consistente en capturar y procesar las partículas nocivas para el organismo. Por así decirlo estas células “se comen y digieren” los restos celulares.
La presentación del antígeno del germen atrapado a otro tipo de leucocito, los linfocitos, para que puedan reconocerlo y así poder eliminarlo.
En condiciones normales, unos niveles de monocitos altos o monocitosis es una condición que suele ser transitoria, así como poco frecuente y nada específica. Unos niveles altos de monocitos deben de ser comprobados en una segunda determinación con analítica sanguínea.
Para definirse como monocitosis el recuento de monocitos debe ser superior a 1.000 unidades por µl y por encima del 10% de manera relativa.
Causas de los monocitos altos
La monocitosis o monocitos altos se define por un aumento en el recuento absoluto de monocitos, con unos niveles por encima de 1.000 unidades por μl. Es característica en el período de recuperación de neutropenias y en convalecencia de cuadros infecciosos.
La monocitosis relativa se puede deber a una baja del resto de cifras de leucocitos, como los neutrófilos o los linfocitos, indicando enfermedades diferentes a una relacionada propiamente con los monocitos.
Las afecciones que pueden causar un aumento de los monocitos en sangre en su fase de resolución son muy variadas y en la mayoría de las ocasiones no indica un trastorno grave. Entre ellas se incluyen:
Infecciones virales (Infección respiratorias, sarampión, mononucleosis, etc.).
Infecciones parasitarias (leishmaniosis y toxoplasmosis).
Enfermedades inflamatorias crónicas.
Patologías autoinmunes.
Tuberculosis.
Sarcoidosis.
Brucelosis.
Listeriosis.
Paludismo.
Enfermedades de la médula ósea.
Unos niveles aumentados de monocitos también pueden indicar la presencia de una hemopatía maligna (leucemias mieloides, linfomas, síndrome mielodisplásico e histiocitosis) y también puede estar asociada a neutropenias crónicas. Este caso es extremadamente infrecuente en comparación con el resto de las causas que pueden elevar la cifra de monocitos.
Los niveles de monocitos también se pueden encontrar disminuidos, siendo esta condición menos frecuente que el aumento de este tipo de células sanguíneas. Las causas pueden ser muy variadas, siendo la más frecuente la infecciosa (tanto viral como bacteriana) y la toma de ciertos fármacos. También puede tener lugar en pacientes que estén en terapia con tratamientos que puedan afectar a la producción celular en la médula ósea, como la quimioterapia o ciertos tratamientos orales.
Para saber el nivel de monocitos en sangre, será necesario la realización de un hemograma o recuento celular sanguíneo a partir de un análisis de sangre.
Valores normales de los monocitos en sangre
Los monocitos suponen normalmente un porcentaje pequeño del total de glóbulos blancos. Si bien los valores normales pueden variar de un paciente a otro, el rango normal de monocitos en sangre oscila entre el 2 y el 10 %.
Ante un aumento llamativo y mantenido de la cifra de monocitos, el médico solicitará un frotis de sangre periférica. Esta prueba consiste en el análisis de la morfología de estas células al microscopio, para comprobar que verdaderamente se trata de monocitos y no de otra célula que pueda simular su forma.
Síntomas de unos monocitos altos o bajos
Esta condición suele ser un hallazgo analítico y normalmente no suelen aparecer síntomas que indiquen la alteración de los niveles de monocitos en la sangre.
Monocitos altos y patologías cardiovasculares
Las enfermedades cardiovasculares y la inflamación crónica de la pared vascular presentan como su principal origen la aparición del proceso de aterosclerosis. Además, la activación de las células del sistema inmunitario está presente en las lesiones ateroscleróticas y favorece su progresión, especialmente los monocitos. Una vez reclutados por el endotelio, gracias a la acción de citoquinas y moléculas de adhesión, adoptan un cambio a un perfil inflamatorio que juega un papel importante en la progresión de la placa aterosclerótica y remodelado miocárdico, origen de problemas cardiovasculares.
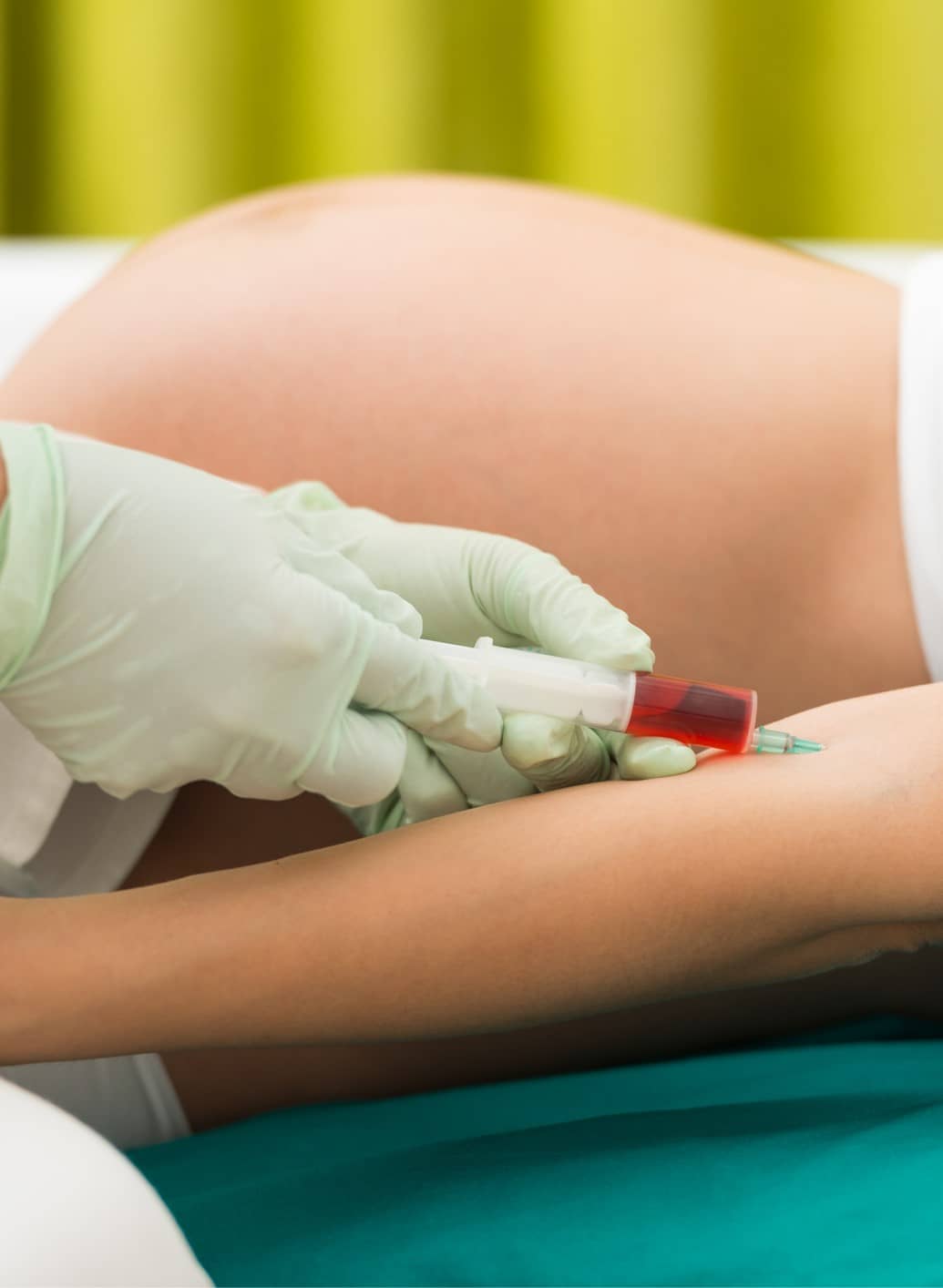
Artículo especializado
Test prenatal Harmony: ¿Qué es y qué detecta?
El test prenatal no invasivo es una prueba que permite evaluar el estado del bebé antes de su nacimiento e identificar posibles anomalías y síndromes genéticos. También permite conocer el sexo del bebé. Estas pruebas son fáciles de realizar e inocuas y permiten identificar aquellas gestantes con fetos que presentan un riesgo elevado de tener una anomalía genética.
Se distinguen diferentes tipos de test no invasivos en función de las anomalías genéticas que son capaces de detectar. El test prenatal Harmony detecta las trisomías más comunes, incluyendo el síndrome de Down, el de Edwards, el de Patau y las anomalías de los cromosomas sexuales. Además, puede detectar el sexo fetal y la opción de detectar el síndrome DiGeorge .
Test prenatales no invasivos
El test Harmony permite la posibilidad de estudiar el ADN libre circulante en el plasma materno para el cribado de las trisomías fetales que, junto con el desarrollo de las técnicas genéticas, nos permiten hoy en día llegar a diagnósticos que antes eran inalcanzables.
Cromosomas y ADN
La información genética o ADN se organiza dentro de cada célula en los cromosomas. Los seres humanos tenemos 23 pares de cromosomas (dos copias de cada par, es decir 46). Los primeros 22 pares se numeran del 1 al 22. El último par determina el sexo. Las niñas tienen dos cromosomas X y los niños un cromosoma X y uno Y. Los problemas de salud y de desarrollo aparecen cuando falta o sobra algún cromosoma o una parte de ellos.
Los test prenatales no invasivos emplean la última tecnología en secuenciación para comparar el ADN fetal respecto al ADN materno, y el riesgo final se calcula aplicando algoritmos en los que se tienen en cuenta la edad materna, la edad gestacional y características del embarazo y la gestante. Todo ello hace que sean precisos y fiables.
Además, pueden incluir otras pruebas para la detección de trisomías relacionadas con abortos espontáneos en el primer trimestre. También son capaces de identificar microdeleciones en las que una pérdida de material genético puede causar la aparición de síndromes genéticos con grandes discapacidades.
También sirven para determinar el sexo fetal y es válido para embarazo gemelar o para donación de óvulos.
Microdeleciones
Las microdeleciones son pequeñas pérdidas de material genético que se asocian con graves problemas del desarrollo en el recién nacido. Es el caso del síndrome de DiGeorge, la microdeleción más frecuente. Este llega a afectar al 1-2% de la población. En él está ausente un pequeño fragmento del cromosoma 22 (22q11.2), lo que acarrea múltiples alteraciones fetales como defectos cardíacos, anomalías en el paladar y la cara, retraso en el desarrollo e inmunodeficiencia, con la aparición de infecciones frecuentes.
¿Cuándo se hace el test Harmony?
Generalmente, el test prenatal Harmony se realizan en la semana 10 del embarazo. Este tipo de prueba destaca por su alta precisión, la tecnología de última generación empleada y la rapidez de los resultados.
Se trata de una prueba no invasiva a partir de una muestra de sangre materna, con lo que desaparece el riesgo para el feto y la madre, siendo totalmente inocuas al contrario que las pruebas invasivas como la biopsia corial o la amniocentesis. Además, permiten un diagnóstico aproximado a más del 99% en las trisomías estudiadas. Su uso ha sido validado en mujeres embarazadas de todas las categorías de edad o riesgo.
Es recomendable que realicen el test Harmony aquellas mujeres gestantes que tienen un elevado riesgo de aneuploidías, considerándose un riesgo elevado cuando se cumple alguna de las siguientes situaciones:
Edad materna avanzada (más de 35 años).
Sospecha ecográfica de aneuploidías.
Historial previo de embarazo con trisomía.
Resultados positivos de cribado prenatal del primer o segundo trimestre.
Existencia de traslocaciones Robertsonianas balanceadas en los progenitores que impliquen a los cromosomas 13 o 21.
Independientemente de los factores de riesgo, esta prueba también está indicada en todos aquellos casos en los que exista ansiedad materna. También se recomienda la realización de pruebas no invasivas en los casos de fecundación in vitro como alternativa sin riesgo al cribado preimplantacional y a la amniocentesis.
¿Que detecta el test Harmony?
El test Harmony es capaz de detectar las siguientes trisomías:
21 (Síndrome de Down).
18 (Síndrome de Edwards).
13 (Síndrome de Patau).
También detecta las aneuploidías en los cromosomas sexuales. Las combinaciones anómalas más comunes son las trisomías XXX (Triple X), XYY (síndrome de Jacobs), XXY (síndrome de Klinefelter), y la monosomía X (síndrome de Turner).
Además, el test Harmony Plus incluye la deleción 22q11.2 (síndrome de DiGeorge).
Finalmente, el test permite averiguar el sexo fetal.
¿Cuándo se recomienda hacer el test prenatal no invasivo?
Los casos en los que se recomienda la realización del test prenatal son:
Mujeres que desean descartar las alteraciones cromosómicas más frecuentes.
Mujeres con riesgo elevado de anomalías cromosómicas tras cribado del primer trimestre (analítica hormonal y ecográfica).
En caso de embarazos anteriores con síndrome de Down.
Mujeres en las que se ha detectado alteraciones ecográficas.
La prueba se puede llevar a cabo a partir del momento en el que haya suficiente material genético fetal circulante en la sangre materna, es decir, a partir de la 9ª semana de embarazo.
¿Para quién está indicado el test Harmony?
El test prenatal no invasivo está indicado para mujeres embarazadas con al menos 10 semanas de gestación, que presenten un embarazo único o gemelar, incluidos embarazos mediante donación de óvulos.
En el caso de embarazo gemelar, únicamente se analizan los cromosomas 21, 18 y 13. De este modo, no se detectan aneuploidías sexuales. En este caso, se podrá averiguar el sexo fetal, si uno de los bebés es niño, o los dos bebés son niñas.
Tipos de test Harmony
Se pueden encontrar 2 tipos de test Harmony, el Test Harmony Medium y el test Harmony Plus:
El test prenatal Harmony Medium consiste en un análisis del ADN fetal que evalúa el riesgo de las trisomías 21 (Síndrome de Down), 18 (Síndrome de Edwards) y 13 (Síndrome de Patau) y los trastornos en los cromosomas sexuales. Las combinaciones anómalas más comunes son las trisomías XXX (Triple X), XYY (síndrome de Jacobs), XXY (síndrome de Klinefelter), y la monosomía X (síndrome de Turner). También ofrece la posibilidad de conocer el sexo fetal.
El test prenatal Harmony Plus incluye las mismas características que el Harmony Medium, junto con la posibilidad de detectar la deleción 22q11.2 (síndrome de DiGeorge).
Toda gestante debe ser informada de la posibilidad de realizarse este diagnóstico no invasivo y de sus características de fiabilidad y posibles alternativas.

Artículo especializado
Test prenatales en embarazo múltiple
Los embarazos gemelares espontáneos alcanzan al 1-2% del total de gestaciones, si bien, en las últimas décadas, han aumentado a un 3-4% en los países desarrollados. Esto es debido al incremento en la edad de la madre por el retraso del momento de la maternidad, así como a las técnicas de reproducción asistida .
El embarazo gemelar es la gestación simultánea de dos fetos en la cavidad uterina, pudiendo ser monocigoto, cuando se origina a partir de un ovulo, o bicigoto, cuando es originado en 2 óvulos. Estos últimos representan el 75% del total en las gestaciones dobles.
En cualquier tipo de embarazo, también en los embarazos múltiples, es importante hacer un seguimiento en el desarrollo del feto (o de los fetos) y comprobar si existe alguna anomalía genética. En la actualidad, los test prenatales no invasivos permiten identificar las anomalías cromosómicas más comunes .
¿Cuándo se recomienda el test prenatal no invasivo en gemelos o mellizos?
En comparación con las gestaciones únicas, las gemelares presentan mayor riesgo de complicaciones, incluyendo defectos congénitos, amenaza de parto prematuro (por la sobre distensión uterina), crecimiento intrauterino restringido (CIR, en los embarazos que tienen una única placenta para los dos embriones), parálisis cerebral y mortalidad cercana al nacimiento. Esto hace que este tipo de embarazos se considere de alto riesgo.
En este sentido, el seguimiento de los embarazos gemelares es similar al de una gestación única, si bien deben ajustarse las revisiones para reforzar su control y descartar la aparición de posibles complicaciones y alteraciones fetales. Se debe actuar para promover un óptimo control del crecimiento fetal y evitar complicaciones frecuentes como la prematuridad.
El diagnóstico ecográfico del embarazo gemelar o múltiple en el primer trimestre es fundamental y, en ocasiones, sobre todo si la primera ecografía transvaginal se realiza muy precozmente, puede haber dudas sobre la presencia real de una gestación múltiple. En el diagnóstico ecográfico del tipo de embarazo múltiple, la zigocidad determina el riesgo de enfermedades genéticas y la corionicidad determina el riesgo de complicaciones perinatales. En un 75 % de los casos existen 2 placentas, una para cada uno de los embriones, mientras que en el 25 % restante existe una placenta compartida, pudiéndose aumentar por ello las alteraciones fetales.
En cuanto a la posible existencia de alguna anomalía cromosómica en alguno de los fetos presentes en el embarazo gemelar, al igual que en los embarazos únicos, existe la posibilidad de realizarse un test prenatal .
Los casos en los que se recomienda la realización del test prenatal no invasivo a partir de un análisis de sangre materna son:
Mujeres que desean descartar las alteraciones cromosómicas más frecuentes.
Mujeres con riesgo elevado de anomalías cromosómicas tras cribado del primer trimestre (analítica hormonal y ecográfica).
En caso de embarazos anteriores con síndrome de Down.
Mujeres en las que se ha detectado alteraciones ecográficas.
La prueba se puede llevar a cabo a partir del momento en el que haya suficiente material genético fetal circulante en la sangre materna, es decir, a partir de la 10 semana de embarazo.
Además, dado que el riesgo de complicaciones tras una amniocentesis gemelar es superior al de un embarazo simple, siempre es recomendable optar previamente por pruebas no invasivas.
¿Cómo se diagnóstica el embarazo múltiple?
Los síntomas más comunes que pueden señalar la presencia de un embarazo múltiple pueden variar en cada mujer. Entre ellos se pueden incluir:
Un útero de tamaño más grande de lo normal.
Más náuseas por la mañana.
Aumento del apetito.
Aumento excesivo del peso materno, en especial al inicio del embarazo.
Percepción de movimientos fetales a la vez en diferentes partes del abdomen.
El diagnóstico de un embarazo múltiple puede producirse al inicio del embarazo. Para ello, además de una historia clínica y un examen físico completo, el diagnóstico se puede obtener mediante las siguientes pruebas:
Análisis de sangre . Los niveles de gonadotropina coriónica humana (hCG) suelen estar mucho más altos en un embarazo múltiple. Además, los niveles de alfa-fetoproteína , proteína liberada por el hígado del feto y que se encuentra en la sangre materna, también pueden estar muy aumentados cuando se trata de un embarazo múltiple.
Ecografía . La técnica por imagen estándar puede permitir identificar la existencia de más de un feto en el embarazo, especialmente si se realiza en las primeras semanas de gestación.
¿Que detecta el test prenatal en embarazo múltiple?
En un embarazo único el test prenatal no invasivo es capaz de detectar diversas alteraciones genéticas congénitas:
Trisomías. En lugar de dos cromosomas, uno de la madre y otro del padre, hay un tercero. Dependiendo del cromosoma que se trate, puede ser trisomía 21 (síndrome de Down), trisomía 18 (síndrome de Edwards) y trisomía 13 (síndrome de Patau).
Anomalías en los cromosomas sexuales. Monosomía X, con un solo cromosoma X (síndrome de Turner), o múltiples cromosomas Y (síndrome de Klinefelter).
Están ausentes pequeños fragmentos cromosómicos, como la deleción de un fragmento del cromosoma 22, el 22q11.2 (síndrome de DiGeorge). Otras microdeleciones dan lugar a otros síndromes, como el de deleción 1p36, el síndrome de Cri-du-chat, el síndrome de Angelman y el síndrome de Prader-Willi.
Triploidías.
Además, el test es capaz de detectar el sexo fetal.
Sin embargo, en embarazos gemelares, el test prenatal no invasivo únicamente incluye la detección de trisomías T13 (síndrome de Patau), T18 (síndrome de Edwards) y T21 (síndrome de Down) y la identificación del sexo fetal.
Los resultados del test en los embarazos de mellizos o gemelos reflejan la probabilidad de que en el embarazo haya al menos un feto afectado. De este modo, informa sobre la presencia de la enfermedad, pero no puede distinguir si se haya presente en uno o en ambos embriones.
En definitiva, el test prenatal no invasivo está indicado para mujeres embarazadas con al menos 10 semanas de gestación, que presenten un embarazo único o gemelar, incluidos embarazos mediante donación de óvulos.
Artículo especializado
¿Qué es la disfagia? Síntomas y tratamiento
Los trastornos que afectan al aparato digestivo y que impiden alimentarse con normalidad pueden derivar en problemas como una alimentación deficiente y la aparición de carencias nutricionales. Una de estas situaciones es la disfagia.
Qué es la disfagia
La disfagia es un trastorno caracterizado por la dificultad en el proceso de deglución de los alimentos bien sea en las fases iniciales ( disfagia orofaríngea ) o la sensación de que existe una obstrucción al paso de los alimentos desde la boca al estómago ( disfagia esofágica ).
La deglución es un proceso neuromuscular complejo que puede verse afectado de forma puntual en cualquier persona sana. También puede ser un proceso crónico en personas con diferentes procesos patológicos como las enfermedades cerebrovasculares y neurodegenerativas o debido al envejecimiento. Estas situaciones impiden al individuo alimentarse correctamente, lo que puede provocar carencias nutricionales y afectación de la calidad de vida.
La disfagia es una patología infradiagnosticada. Algunos autores estiman que afecta a cerca de 2 millones de españoles, de los cuales solo un 10 % está correctamente diagnosticado y tratado.
El proceso de la deglución
La deglución tiene un papel clave en la alimentación del individuo y resulta la puerta de entrada a los procesos fisiológicos de digestión, absorción y transformación de los nutrientes que necesita el organismo.
El inicio voluntario de la deglución requiere la unión de múltiples estímulos sensoriales originados en la orofaringe, los cuales se dirigen a la médula espinal y la corteza cerebral. La deglución normal ocurre en cuatro grandes fases. Cuando alguna de estas fases se ve afectada se origina la disfagia:
La fase oral preparatoria está bajo control voluntario y su objetivo es la masticación y la formación del bolo.
La fase oral propulsiva es también voluntaria y se caracteriza por la propulsión del bolo por acción de la lengua y los movimientos musculares coordinados en la orofarínge.
La fase faríngea es involuntaria. En ella los diferentes estímulos como el sabor, el olor, la textura de los alimentos, que envían información al sistema nervioso central, originan un patrón deglutorio faríngeo caracterizado por una secuencia coordinada de acontecimientos motores que ocasionan el cierre de la nasofaringe y de la vía respiratoria y la apertura del esfínter esofágico superior, con contracción de los músculos constrictores faríngeos.
La fase esofágica se inicia con la apertura del esfínter esofágico superior seguido de la peristalsis esofágica, que permite el descenso del bolo alimenticio al estómago.
Factores de riesgo de la disfagia
Los factores de riesgo que se relacionan con la aparición de disfagia son muy variables, e incluyen el envejecimiento, las enfermedades mentales, neurológicas y los eventos cerebrovasculares, las afecciones musculares y las alteraciones anatómicas.
En personas de edad avanzada algunos factores como la perdida de dientes, prótesis móviles, atrofia de músculos masticatorios y la menor producción de saliva inciden negativamente en el proceso de las primeras fases de la deglución.
La presencia de compresiones extrínsecas, alteraciones anatómicas y cirugías previas o la radioterapia de tumores o lesiones faríngeas, laríngeas o maxilofaciales también son factores de riesgo para disfagia.
Por otro lado, la presencia de disfagia ha sido asociada con un aumento del riesgo de sufrir complicaciones pulmonares, infecciones respiratorias y de la mortalidad. De ahí que el diagnóstico precoz y el manejo multidisciplinario de la disfagia en pacientes con daño cerebral y/o institucionalizados sea necesario.
Causas de la disfagia
Es importante distinguir entre las causas que provocan disfagia orofaríngea y las que provocan disfagia esofágica.
Disfagia orofaríngea
En los pacientes jóvenes, la disfagia orofaríngea generalmente es provocada por enfermedades musculares, membranas o anillos. En los individuos de más edad habitualmente es provocada por trastornos del sistema nervioso central, como el accidente cerebrovascular, enfermedad de Parkinson y demencia. El envejecimiento normal puede provocar anomalías leves de la motilidad esofágica, rara vez sintomáticas.
Entre las causas mecánicas y obstructivas están las infecciones, el divertículo de Zenker, miopatías (miositis, fibrosis, barra cricofaríngea), neoplasias de cabeza y cuello y consecuencias de intervenciones quirúrgicas y/o radioterapéuticas sobre estos tumores, entre otras.
Los trastornos neuromusculares y patologías del sistema nervioso central como parálisis de pares craneanos o parálisis bulbar, esclerosis múltiple, esclerosis lateral amioatrófica, miastenia gravis, entre otros, también pueden ser motivo de disfagia.
Disfagia esofágica
Existen causas obstructivas de la luz del esófago que ocasionan disfagia por invasión directa, compresión, o linfadenomegalias. Los tumores compresivos, como el cáncer de pulmón, linfoma, neoplasias de esófago, estenosis péptica secundaria a enfermedad por reflujo gastroesofágico y anillos y membranas esofágicas.
También las enfermedades de la mucosa que ocasionan inflamación y/o fibrosis. Entre estas tenemos las infecciones, esofagitis por cáusticos, píldoras, radiaciones, esofagitis eosinofílica y las enfermedades neuromusculares como acalasia, esclerodermia, enfermedades mixtas del tejido conectivo (miositis) y espamo esofágico difuso, entre otras.
Tipos de disfagia
Disfagia aguda
La disfagia aguda se caracteriza por la imposibilidad repentina para la deglución, casi siempre relacionada con la impactación de cuerpos extraños, como trozos de alimentos en la luz del esófago. por lo que requiere una atención médica inmediata.
Disfagia no aguda
Puede ser tanto orofaríngea como esofágica, de evolución crónica, progresiva o intermitente, dependiendo de la causa.
Disfagia funcional
Se caracteriza por tener la sensación de un tránsito anormal del bolo a través del esófago. Está frecuentemente relacionado con la ansiedad y estrés.
Este tipo de disfagia suele ser intermitente y mejora con el tiempo. Se recomienda modificaciones en el estilo de vida como comer despacio y masticar bien. En casos de intensidad notable, pueden indicarse fármacos ansiolíticos y antidepresivos.
Síntomas de la disfagia
La manifestación de los síntomas permite identificar el tipo de disfagia en la gran mayoría de los casos, aunque los síntomas en ocasiones se solapan.
Los principales síntomas y manifestaciones observables en la disfagia orofaríngea son:
Salivación o babeo.
Presencia de restos de comida en la boca.
Síntomas de aspiración como tos frecuente durante la deglución y sensación de ahogo.
Regurgitación nasal u oral inmediata.
Dolor al tragar.
Dificultad para la masticación.
Deglución fraccionada y repetida.
En la disfagia esofágica los síntomas son principalmente sensación de atragantamiento en el tórax, dolor torácico, que puede acompañarse de regurgitación y ardor justo detrás del esternón.
También pueden aparecer síntomas asociados con la pérdida de peso, la desnutrición, la deshidratación y las infecciones respiratorias frecuentes.
Tratamiento de la disfagia
Es muy importante una detección precoz de la disfagia para establecer las pautas de alimentación y reeducación del patrón deglutorio. Las medidas generales de rehabilitación deglutoria incluyen:
Acompañamiento de personas con disfagia durante la ingesta, en un ambiente tranquilo, sin ruidos ni distracciones, bien iluminado, con el tiempo suficiente por los posibles déficits motores y el retraso del reflejo deglutorio. Es importante también realizar una buena limpieza bucal antes y después de comer.
Estrategias posturales para evitar la broncoaspiración. La flexión cervical anterior es una de las más seguras, consistente en aproximar el mentón al esternón, sin hiperflexión.
Las estrategias de incremento sensorial oral son útiles cuando hay alteraciones de la sensibilidad oral. Estas incluyen la estimulación mecánica de la lengua y de los pilares faríngeos (presión de la cuchara al introducir el alimento), el uso de alimentos fríos y ácidos (limón/lima), y las modificaciones del volumen y sabor del bolo.
Ejercicios de biofeedback y reeducación muscular que mejoran la fisiología de la deglución incrementando el tono, la sensibilidad y motricidad de las estructuras que intervienen en ella.
Maniobras deglutorias especificas indicadas para mejorar la eficacia y seguridad de la deglución, favoreciendo el cierre laríngeo y el paso del bolo al esófago sin residuos en personas con un buen estado cognitivo y que pueda colaborar activamente.
Por otro lado, es indispensable abordar la causa que provoca la disfagia, requiriendo de tratamiento médico adecuado (como en las esofagitis pépticas, eosinofílicas e infecciones) y en algunos casos el uso de técnicas quirúrgicas apropiadas.
Finalmente, también es fundamental el tratamiento dietético-nutricional individualizado cuya finalidad es reducir el riesgo de atragantamiento mediante la adaptación del volumen y viscosidad de los líquidos y de las texturas de los sólidos para convertirlos en purés o hacerlos fácilmente masticables. Además, se deben cubrir todas las necesidades nutricionales a partir de los alimentos y líquidos. En ocasiones puede estar indicada la suplementación nutricional o la consideración de vías de nutrición alternativas a la vía oral.
¿Puedo confiar en Savia?
Todos nuestros servicios están creados con la intención de acercar la atención médica y sanitaria de calidad a todos los trabajadores y sus familias, siempre con la garantía de MAPFRE. Queremos establecer una relación de total confianza.